Molecular Analysis of 55 Spanish Patients with Acute Intermittent Porphyria

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Biochemical Determinations
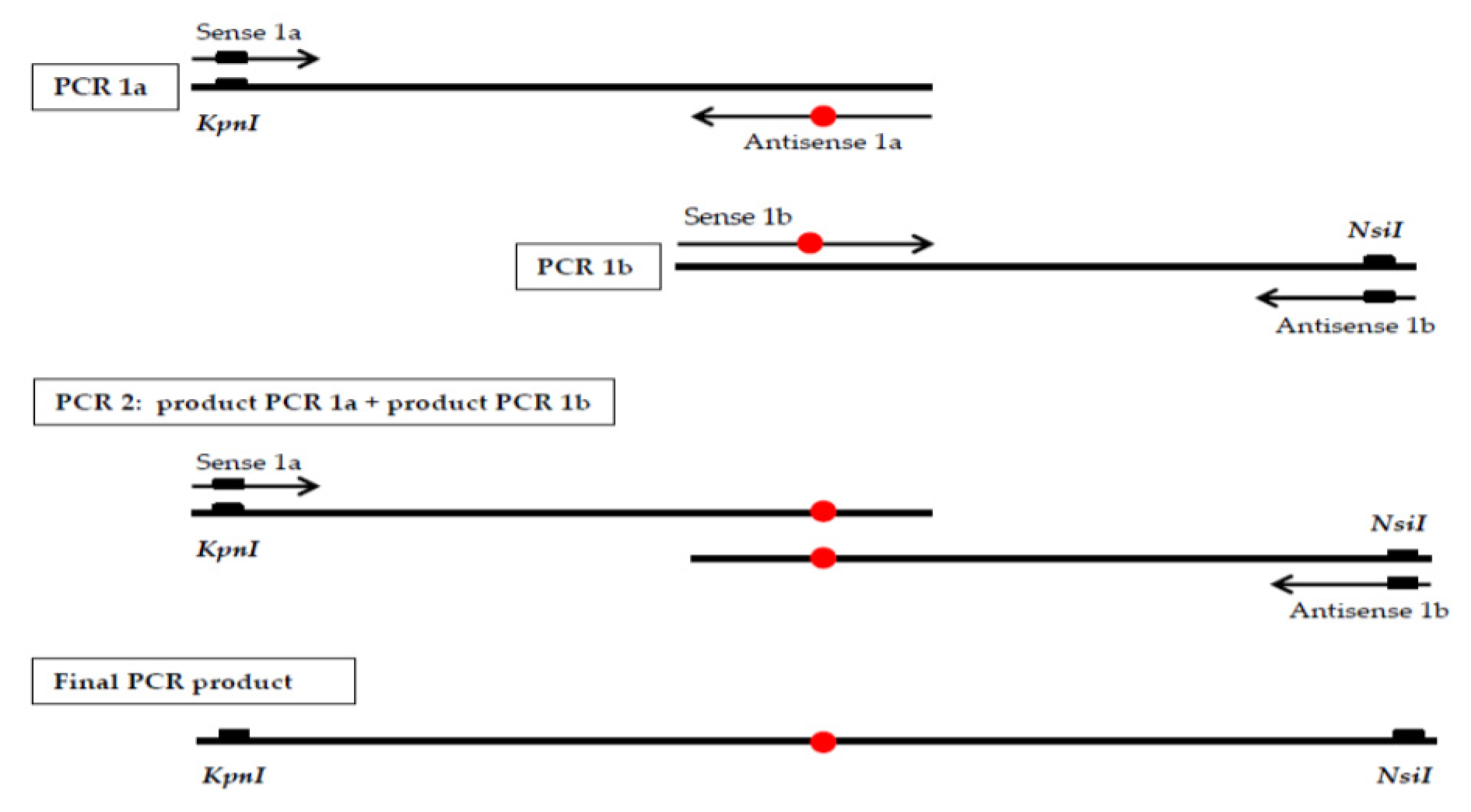
2.2. DNA Analysis
2.3. RNA Analysis
2.4. Prokaryotic Expression and Characterization of the Novel HMBS Missense Mutation
3. Results
3.1. Patients
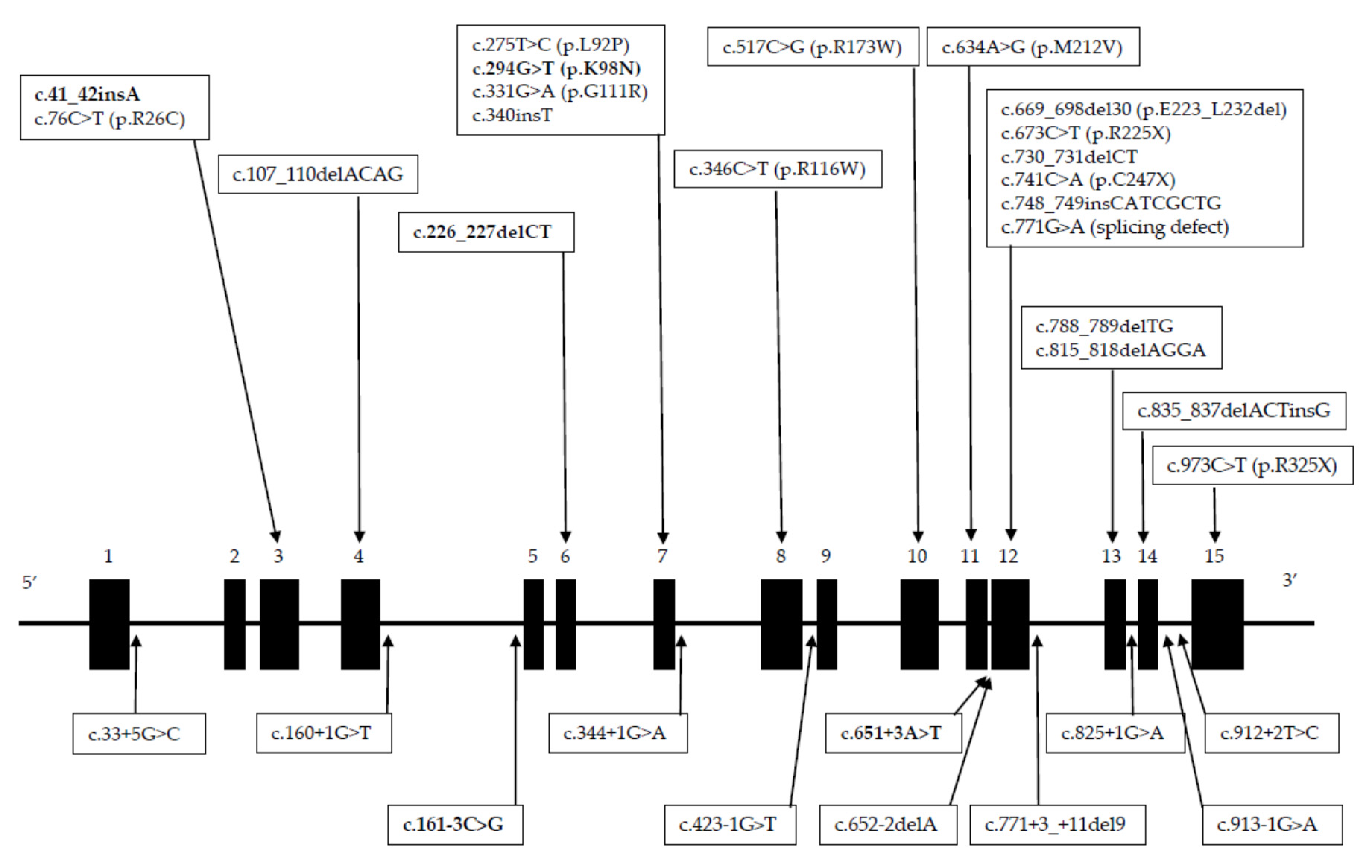
3.2. Mutations Identified
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderson, K.E.; Sassa, S.; Bishop, D.F.; Desnick, R.J. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In The Metabolic and Molecular Basis of Inherited Disease; Scriver, C.R., Beaudet, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2991–3062. [Google Scholar]
- Gill, R.; Kolstoe, S.E.; Mohammed, F.; Al, D.B.A.; Mosely, J.E.; Sarwar, M.; Cooper, J.B.; Wood, S.P.; Shoolingin-Jordan, P.M. Structure of human porphobilinogen deaminase at 2.8 A: The molecular basis of acute intermittent porphyria. Biochem. J. 2009, 420, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Bung, N.; Roy, A.; Chen, B.; Das, D.; Pradhan, M.; Yasuda, M.; New, M.I.; Desnick, R.J.; Bulusu, G. Human hydroxymethylbilane synthase: Molecular dynamics of the pyrrole chain elongation identifies step-specific residues that cause AIP. Proc. Natl. Acad. Sci. USA 2018, 115, E4071–E4080. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Maddukuri, V.C.; Yazici, C.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.R.; Phillips, J.D.; Naik, H.; Peter, I.; Baillargeon, G.; et al. Acute porphyrias in the USA: Features of 108 subjects from porphyrias consortium. Am. J. Med. 2014, 127, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Lamon, J.; With, T.K.; Redeker, A.G. The Hoesch test: Bedside screening for urinary porphobilinogen in patients with suspected porphyria. Clin. Chem. 1974, 20, 1438–1440. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.H.; Smith, S.G.; Smyth, S.J. Laboratory investigation of the porphyrias. Ann. Clin. Biochem. 1990, 27 Pt 5, 395–412. [Google Scholar] [CrossRef]
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 2005, 142, 439–450. [Google Scholar] [CrossRef]
- Namba, H.; Narahara, K.; Tsuji, K.; Yokoyama, Y.; Seino, Y. Assignment of human porphobilinogen deaminase to 11q24.1----q24.2 by in situ hybridization and gene dosage studies. Cytogenet. Cell Genet. 1991, 57, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.W.; Warner, C.A.; Chen, C.H.; Desnick, R.J. Hydroxymethylbilane synthase: Complete genomic sequence and amplifiable polymorphisms in the human gene. Genomics 1993, 15, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Chretien, S.; Dubart, A.; Beaupain, D.; Raich, N.; Grandchamp, B.; Rosa, J.; Goossens, M.; Romeo, P.H. Alternative transcription and splicing of the human porphobilinogen deaminase gene result either in tissue-specific or in housekeeping expression. Proc. Natl. Acad. Sci. USA 1988, 85, 6–10. [Google Scholar] [CrossRef]
- Grandchamp, B.; De Verneuil, H.; Beaumont, C.; Chretien, S.; Walter, O.; Nordmann, Y. Tissue-specific expression of porphobilinogen deaminase. Two isoenzymes from a single gene. Eur. J. Biochem. 1987, 162, 105–110. [Google Scholar] [CrossRef]
- Song, G.; Li, Y.; Cheng, C.; Zhao, Y.; Gao, A.; Zhang, R.; Joachimiak, A.; Shaw, N.; Liu, Z.J. Structural insight into acute intermittent porphyria. FASEB J. 2009, 23, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Mustajoki, P. Normal erythrocyte uroporphyrinogen I synthase in a kindred with acute intermittent porphyria. Ann. Intern. Med. 1981, 95, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, Y.; Puy, H. Human hereditary hepatic porphyrias. Clin. Chim. Acta 2002, 325, 17–37. [Google Scholar] [CrossRef]
- Sassa, S. Modern diagnosis and management of the porphyrias. Br. J. Haematol. 2006, 135, 281–292. [Google Scholar] [CrossRef]
- Puy, H.; Deybach, J.C.; Lamoril, J.; Robreau, A.M.; Da Silva, V.; Gouya, L.; Grandchamp, B.; Nordmann, Y. Molecular epidemiology and diagnosis of PBG deaminase gene defects in acute intermittent porphyria. Am. J. Hum. Genet. 1997, 60, 1373–1383. [Google Scholar] [CrossRef]
- Mendez, M.; Moran-Jimenez, M.J.; Gomez-Abecia, S.; Garcia-Bravo, M.; Garrido-Astray, M.C.; Fontanellas, A.; Poblete-Gutierrez, P.; Frank, J.; Enriquez de Salamanca, R. Identification and characterization of HMBS gene mutations in Spanish patients with acute intermittent porphyria. Cell Mol. Biol. 2009, 55, 55–63. [Google Scholar]
- Cormack, B. Mutagenesis of cloned DNA. In Current Protocols in Molecular Biology; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; John Wiley & sons Inc.: New York, NY, USA, 2003; pp. 2851–8510. [Google Scholar]
- Puy, H.; Gross, U.; Deybach, J.C.; Robreau, A.M.; Frank, M.; Nordmann, Y.; Doss, M. Exon 1 donor splice site mutations in the porphobilinogen deaminase gene in the non-erythroid variant form of acute intermittent porphyria. Hum. Genet. 1998, 103, 570–575. [Google Scholar] [CrossRef]
- Kauppinen, R.; Mustajoki, S.; Pihlaja, H.; Peltonen, L.; Mustajoki, P. Acute intermittent porphyria in Finland: 19 mutations in the porphobilinogen deaminase gene. Hum. Mol. Genet. 1995, 4, 215–222. [Google Scholar] [CrossRef]
- Chen, B.; Solis-Villa, C.; Erwin, A.L.; Balwani, M.; Nazarenko, I.; Phillips, J.D.; Desnick, R.J.; Yasuda, M. Identification and characterization of 40 novel hydroxymethylbilane synthase mutations that cause acute intermittent porphyria. J. Inherit. Metab. Dis. 2019, 42, 186–194. [Google Scholar] [CrossRef]
- Floderus, Y.; Shoolingin-Jordan, P.M.; Harper, P. Acute intermittent porphyria in Sweden. Molecular, functional and clinical consequences of some new mutations found in the porphobilinogen deaminase gene. Clin. Genet. 2002, 62, 288–297. [Google Scholar] [CrossRef]
- Gu, X.F.; de Rooij, F.; de Baar, E.; Bruyland, M.; Lissens, W.; Nordmann, Y.; Grandchamp, B. Two novel mutations of the porphobilinogen deaminase gene in acute intermittent porphyria. Hum. Mol. Genet. 1993, 2, 1735–1736. [Google Scholar] [CrossRef] [PubMed]
- Whatley, S.D.; Woolf, J.R.; Elder, G.H. Comparison of complementary and genomic DNA sequencing for the detection of mutations in the HMBS gene in British patients with acute intermittent porphyria: Identification of 25 novel mutations. Hum. Genet. 1999, 104, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Robreau-Fraolini, A.M.; Puy, H.; Aquaron, C.; Bogard, C.; Traore, M.; Nordmann, Y.; Aquaron, R.; Deybach, J.C. Porphobilinogen deaminase gene in African and Afro-Caribbean ethnic groups: Mutations causing acute intermittent porphyria and specific intragenic polymorphisms. Hum. Genet. 2000, 107, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.F.; de Rooij, F.; Lee, J.S.; Te Velde, K.; Deybach, J.C.; Nordmann, Y.; Grandchamp, B. High prevalence of a point mutation in the porphobilinogen deaminase gene in Dutch patients with acute intermittent porphyria. Hum. Genet. 1993, 91, 128–130. [Google Scholar] [CrossRef]
- De Siervi, A.; Mendez, M.; Parera, V.E.; Varela, L.; Batlle, A.M.; Rossetti, M.V. Acute intermittent porphyria: Characterization of two novel mutations in the porphobilinogen deaminase gene, one amino acid deletion (453-455delAGC) and one splicing aceptor site mutation (IVS8-1G>T). Hum. Mutat. 1999, 14, 355. [Google Scholar] [CrossRef]
- Mgone, C.S.; Lanyon, W.G.; Moore, M.R.; Louie, G.V.; Connor, J.M. Identification of five novel mutations in the porphobilinogen deaminase gene. Hum. Mol. Genet. 1994, 3, 809–811. [Google Scholar] [CrossRef]
- Solis, C.; Lopez-Echaniz, I.; Sefarty-Graneda, D.; Astrin, K.H.; Desnick, R.J. Identification and expression of mutations in the hydroxymethylbilane synthase gene causing acute intermittent porphyria (AIP). Mol. Med. 1999, 5, 664–671. [Google Scholar] [CrossRef]
- Martinez di Montemuros, F.; Di Pierro, E.; Biolcati, G.; Rocchi, E.; Bissolotti, E.; Tavazzi, D.; Fiorelli, G.; Cappellini, M.D. Acute intermittent porphyria: Heterogeneity of mutations in the hydroxymethylbilane synthase gene in Italy. Blood Cells Mol. Dis. 2001, 27, 961–970. [Google Scholar] [CrossRef]
- Guillen-Navarro, E.; Carbonell, P.; Glover, G.; Sanchez-Solis, M.; Fernandez-Barreiro, A. Novel HMBS founder mutation and significant intronic polymorphism in Spanish patients with acute intermittent porphyria. Ann. Hum. Genet. 2004, 68, 509–514. [Google Scholar] [CrossRef]
- Mgone, C.S.; Lanyon, W.G.; Moore, M.R.; Louie, G.V.; Connor, J.M. Detection of a high mutation frequency in exon 12 of the porphobilinogen deaminase gene in patients with acute intermittent porphyria. Hum. Genet. 1993, 92, 619–622. [Google Scholar] [CrossRef]
- Whatley, S.D.; Mason, N.G.; Woolf, J.R.; Newcombe, R.G.; Elder, G.H.; Badminton, M.N. Diagnostic strategies for autosomal dominant acute porphyrias: Retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin. Chem. 2009, 55, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Grandchamp, B.; Picat, C.; de Rooij, F.; Beaumont, C.; Wilson, P.; Deybach, J.C.; Nordmann, Y. A point mutation G----A in exon 12 of the porphobilinogen deaminase gene results in exon skipping and is responsible for acute intermittent porphyria. Nucleic Acids Res. 1989, 17, 6637–6649. [Google Scholar] [CrossRef] [PubMed]
- De Siervi, A.; Rossetti, M.V.; Parera, V.E.; Astrin, K.H.; Aizencang, G.I.; Glass, I.A.; Batlle, A.M.; Desnick, R.J. Identification and characterization of hydroxymethylbilane synthase mutations causing acute intermittent porphyria: Evidence for an ancestral founder of the common G111R mutation. Am. J. Med. Genet. 1999, 86, 366–375. [Google Scholar] [CrossRef]
- Llewellyn, D.H.; Scobie, G.A.; Urquhart, A.J.; Whatley, S.D.; Roberts, A.G.; Harrison, P.R.; Elder, G.H. Acute intermittent porphyria caused by defective splicing of porphobilinogen deaminase RNA: A synonymous codon mutation at -22 bp from the 5′ splice site causes skipping of exon 3. J. Med. Genet. 1996, 33, 437–438. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Surin, V.L.; Luchinina Iu, A.; Selivanova, D.S.; Pustovoit Ia, S.; Karpova, I.S.; Pivnik, A.V.; Luk’ianenko, A.V.; Kravchenko, S.K. Molecular genetic study of acute intermittent porphyria in Russia: Mutation analysis and functional polymorphism search in porphobilinogen deaminase gene. Genetika 2010, 46, 540–552. [Google Scholar] [CrossRef]
- Cerbino, G.N.; Gerez, E.N.; Varela, L.S.; Melito, V.A.; Parera, V.E.; Batlle, A.; Rossetti, M.V. Acute intermittent porphyria in Argentina: An update. Biomed. Res. Int. 2015, 2015, 946387. [Google Scholar] [CrossRef]
- Petersen, N.E.; Nissen, H.; Hansen, T.S.; Rasmussen, K.; Brock, A.; Horder, M. R325X mutation in exon 15 of the hydroxymethylbilane synthase gene identified in two Danish families with acute intermittent porphyria. Clin. Chem. 1996, 42, 106–107. [Google Scholar] [CrossRef]
- Hentze, M.W.; Kulozik, A.E. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 1999, 96, 307–310. [Google Scholar] [CrossRef]
- Maquat, L.E. Defects in RNA splicing and the consequence of shortened translational reading frames. Am. J. Hum. Genet. 1996, 59, 279–286. [Google Scholar]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000, 28, 4364–4375. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient (Sex) Age 1 | ALA (mg/L) n.r. ≤ 7.5 | PBG (mg/L) n.r. ≤ 2.5 | Total Porphyrins (µg/g Creatinine) n.r. ≤ 200 | HMBS Activity 2 | Mutation 3 | Location | Reference |
|---|---|---|---|---|---|---|---|
| P1 (F) 22 | 60 | 47 | 22,025 | 105 | c.33+5G > C (splicing defect) 4 | Intron 1 | [19] |
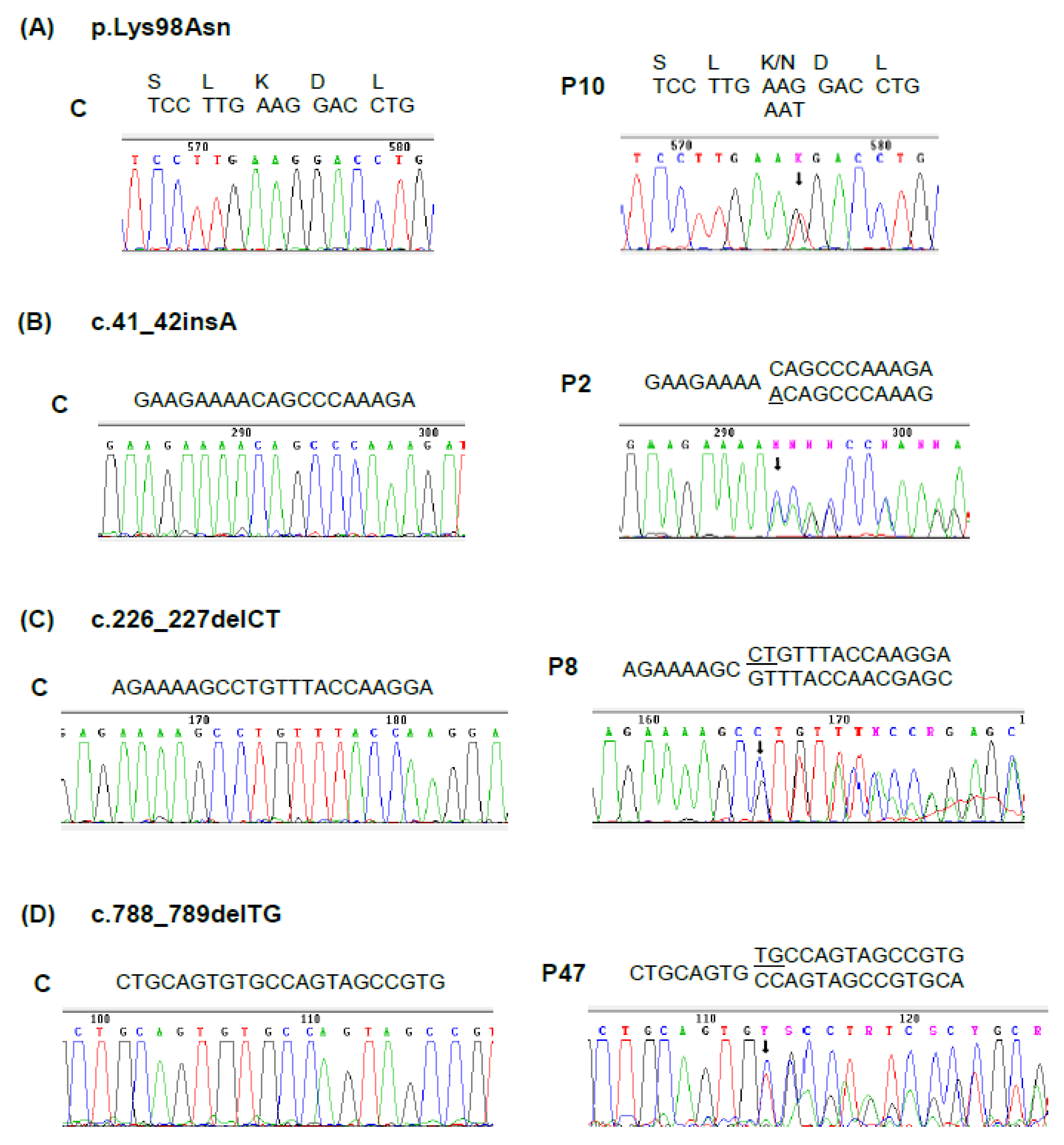
| P2 (F) 33 | 13 | 21 | 435 | 48 | c.41_42insA | Exon 3 | This study |
| P3 (F) 29 | 114 | 104 | 1120 | 52 | c.76C > T (p.R26C) | Exon 3 | [20] |
| P4 (F) 26 | 9 | 12 | 716 | 33 | |||
| P5 (F) 23 | 9 | 10 | 364 | 69 | c.107_110delACAG 4 | Exon 4 | [21] |
| P6 (F) 28 | 8 | 11 | 993 | 55 | c.160 + 1G > T (splicing defect) 4 | Intron 4 | [16] |
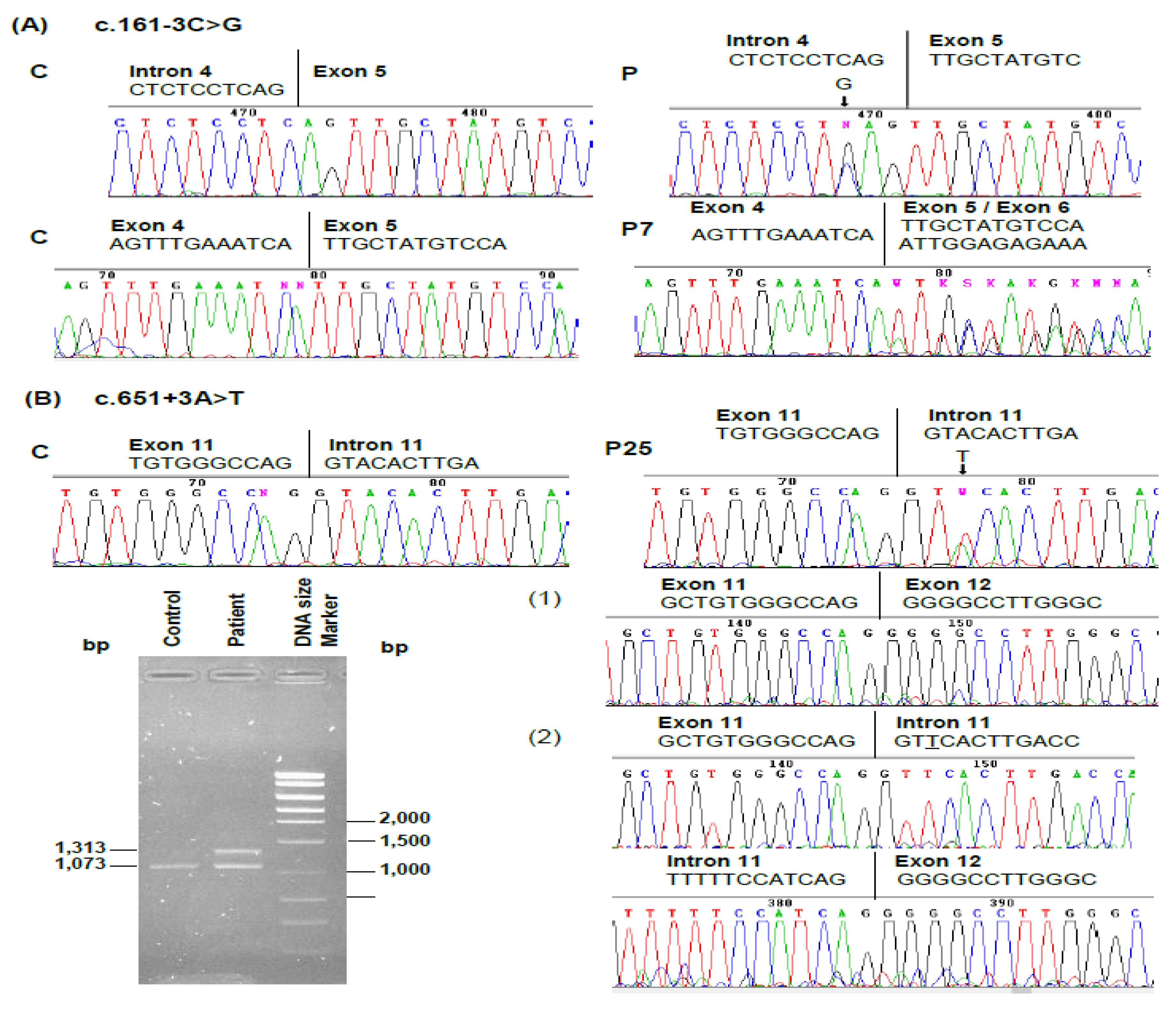
| P7 (F) 45 | 14 | 39 | 234 | 62 | c.161–3C > G (splicing defect) | Intron 4 | This study |
| P8 (F) 38 | n.a. | n.a. | n.a. | 75 | c.226_227delCT | Exon 6 | This study |
| P9 (M) 62 | n.a. | n.a. | n.a. | 53 | c.275T > C (p.L92P) | Exon 7 | [22] |
| P10 (F) 34 | 57 | 151 | 1926 | 61 | c.294G > T (p.K98N) | Exon 7 | This study |
| P11 (F) 29 | n.a. | n.a. | n.a. | 47 | c.331G > A (p.G111R) | Exon 7 | [23] |
| P12 (F) 61 | 20 | 35 | n.a. | 72 | |||
| P13 (F) 32 | n.a. | n.a. | n.a. | 50 | c.340insT | Exon 7 | [24] |
| P14 (F) 38 | 73 | 21 | 23,381 | 57 | |||
| P15 (F) 21 | 37 | 41 | 1232 | 60 | |||
| P16 (F) 28 | n.a. | n.a. | n.a. | 46 | c.344 + 1G > A (splicing defect) 4 | Intron 7 | [25] |
| P17 (F) 46 | 67 | 45 | 910 | 60 | c.346C > T (p.R116W) | Exon 8 | [26] |
| P18 (F) 19 | 17 | 47 | 392 | 48 | |||
| P19 (M) 25 | n.a. | n.a. | n.a. | 44 | |||
| P20 (F) 26 | 14 | 21 | 459 | 46 | c.423–1G > T (splicing defect) 4 | Intron 8 | [27] |
| P21 (F) 34 | 25 | 50 | 794 | 44 | c.517C > G (p.R173W) | Exon 10 | [28] |
| P22 (F) 22 | 47 | 25 | 381 | 59 | |||
| P23 (F) 28 | 14 | 71 | 720 | 55 | |||
| P24 (F) 51 | n.a. | n.a. | n.a. | 70 | c.634A > G (p.M212V) | Exon 11 | [29] |
| P25 (F) 17 | 127 | 143 | 527 | 59 | c.651 + 3A > T (splicing defect) | Intron 11 | This study |
| P26 (F) 27 | 55 | 74 | n.a. | 58 | c.652–2delA (splicing defect) | Intron 11 | [30] |
| P27 (F) 43 | 10 | 5 | 669 | 71 | c.669_698del30 (p.E223_L232del) | Exon 12 | [31] |
| P28 (F) 26 | 30 | 7 | 316 | 55 | |||
| P29 (F) 36 | 38 | 156 | 1403 | 52 | |||
| P30 (F) 30 | 83 | 235 | 127 | 49 | |||
| P31 (M) 40 | 28 | 66 | 374 | 56 | |||
| P32 (F) 35 | n.a. | n.a. | 1578 | 44 | |||
| P33 (F) 56 | n.a. | n.a. | n.a. | 24 | |||
| P34 (F) 37 | n.a. | n.a. | n.a. | 54 | |||
| P35 (M) 38 | n.a. | n.a. | 358 | 58 | |||
| P36 (F) 41 | 10 | 4 | 908 | 64 | |||
| P37 (F) 62 | n.a. | n.a. | n.a. | 48 | |||
| P38 (F) 25 | 125 | 109 | 794 | 64 | c.673C > T (p.R225X) | Exon 12 | [20] |
| P39 (M) 56 | n.a. | n.a. | n.a. | 61 | c.730_731delCT 4 | Exon 12 | [32] |
| P40 (F) 28 | n.a. | n.a. | n.a. | 60 | |||
| P41 (F) 49 | 55 | 103 | 1115 | 55 | c.741C > A (p.C247X) 4 | Exon 12 | [24] |
| P42 (F) 44 | 31 | 31 | 524 | 65 | c.748_749insCATCGCTG 4 | Exon 12 | [33] |
| P43 (M) 37 | n.a. | n.a. | n.a. | 45 | c.771G > A (splicing defect) 4 | Exon12 | [34] |
| P44 (F) 57 | n.a. | n.a. | n.a. | 57 | c.771 + 3_ +11del9 (splicing defect) | Intron 12 | [17] |
| P45 (F) 36 | 21 | 28 | 801 | 48 | |||
| P46 (F) 20 | 13 | 26 | 973 | 55 | |||
| P47 (M) 42 | 41 | 124 | 608 | 65 | c.788_789delTG | Exon 13 | This study |
| P48 (F) 41 | 19 | 68 | 11,258 | 55 | c.815_818delAGGA 4 | Exon 13 | [35] |
| P49 (F) 23 | 37 | 62 | 887 | 74 | c.825 + 1G > A (splicing defect) 4 | Intron 13 | [36] |
| P50 (F) 28 | n.a. | n.a. | n.a. | 61 | |||
| P51 (F) 28 | 12 | 32 | 246 | 68 | c.835_837delACTinsG | Exon 14 | [29] |
| P52 (F) 39 | 9 | 13 | 498 | 60 | c.912 + 2T > C (splicing defect) 4 | Intron 14 | [37] |
| P53 (F) 16 | 43 | 89 | 992 | 50 | c.913–1G > A (splicing defect) 4 | Intron 14 | [38] |
| P54 (M) 38 | 20 | 23 | 2264 | 53 | c.973C > T (p.R325X) 4 | Exon 15 | [39] |
| P55 (F) 30 | 38 | 146 | 1266 | 78 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morán-Jiménez, M.-J.; Borrero-Corte, M.-J.; Jara-Rubio, F.; García-Pastor, I.; Díaz-Díaz, S.; Castelbón-Fernandez, F.-J.; Enríquez-de-Salamanca, R.; Méndez, M. Molecular Analysis of 55 Spanish Patients with Acute Intermittent Porphyria. Genes 2020, 11, 924. https://doi.org/10.3390/genes11080924
Morán-Jiménez M-J, Borrero-Corte M-J, Jara-Rubio F, García-Pastor I, Díaz-Díaz S, Castelbón-Fernandez F-J, Enríquez-de-Salamanca R, Méndez M. Molecular Analysis of 55 Spanish Patients with Acute Intermittent Porphyria. Genes. 2020; 11(8):924. https://doi.org/10.3390/genes11080924
Chicago/Turabian StyleMorán-Jiménez, María-José, María-José Borrero-Corte, Fátima Jara-Rubio, Inmaculada García-Pastor, Silvia Díaz-Díaz, Francisco-Javier Castelbón-Fernandez, Rafael Enríquez-de-Salamanca, and Manuel Méndez. 2020. "Molecular Analysis of 55 Spanish Patients with Acute Intermittent Porphyria" Genes 11, no. 8: 924. https://doi.org/10.3390/genes11080924
APA StyleMorán-Jiménez, M.-J., Borrero-Corte, M.-J., Jara-Rubio, F., García-Pastor, I., Díaz-Díaz, S., Castelbón-Fernandez, F.-J., Enríquez-de-Salamanca, R., & Méndez, M. (2020). Molecular Analysis of 55 Spanish Patients with Acute Intermittent Porphyria. Genes, 11(8), 924. https://doi.org/10.3390/genes11080924