Progesterone Receptor Coregulators as Factors Supporting the Function of the Corpus Luteum in Cows


Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Collection
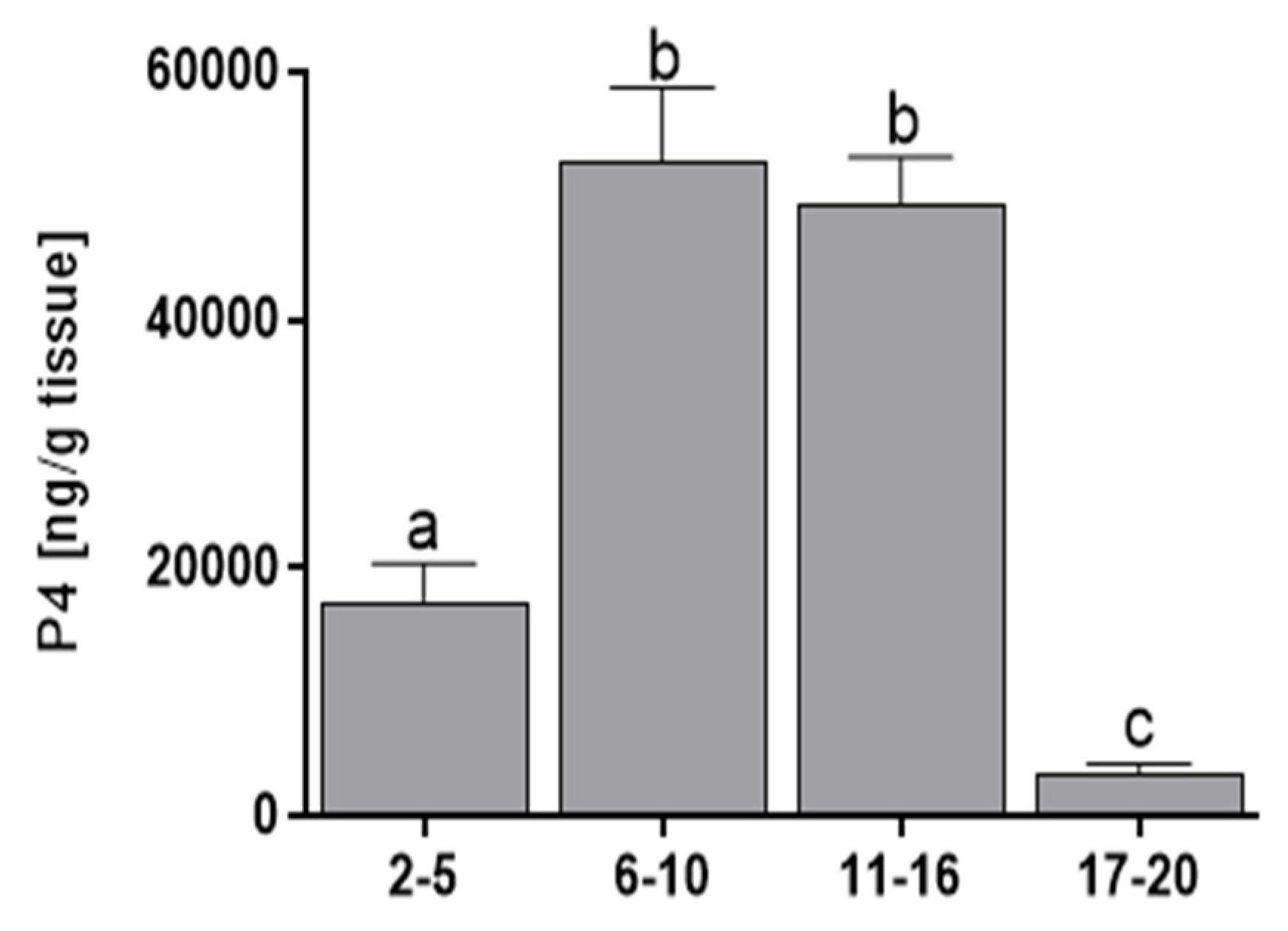
2.2. Progesterone Determination
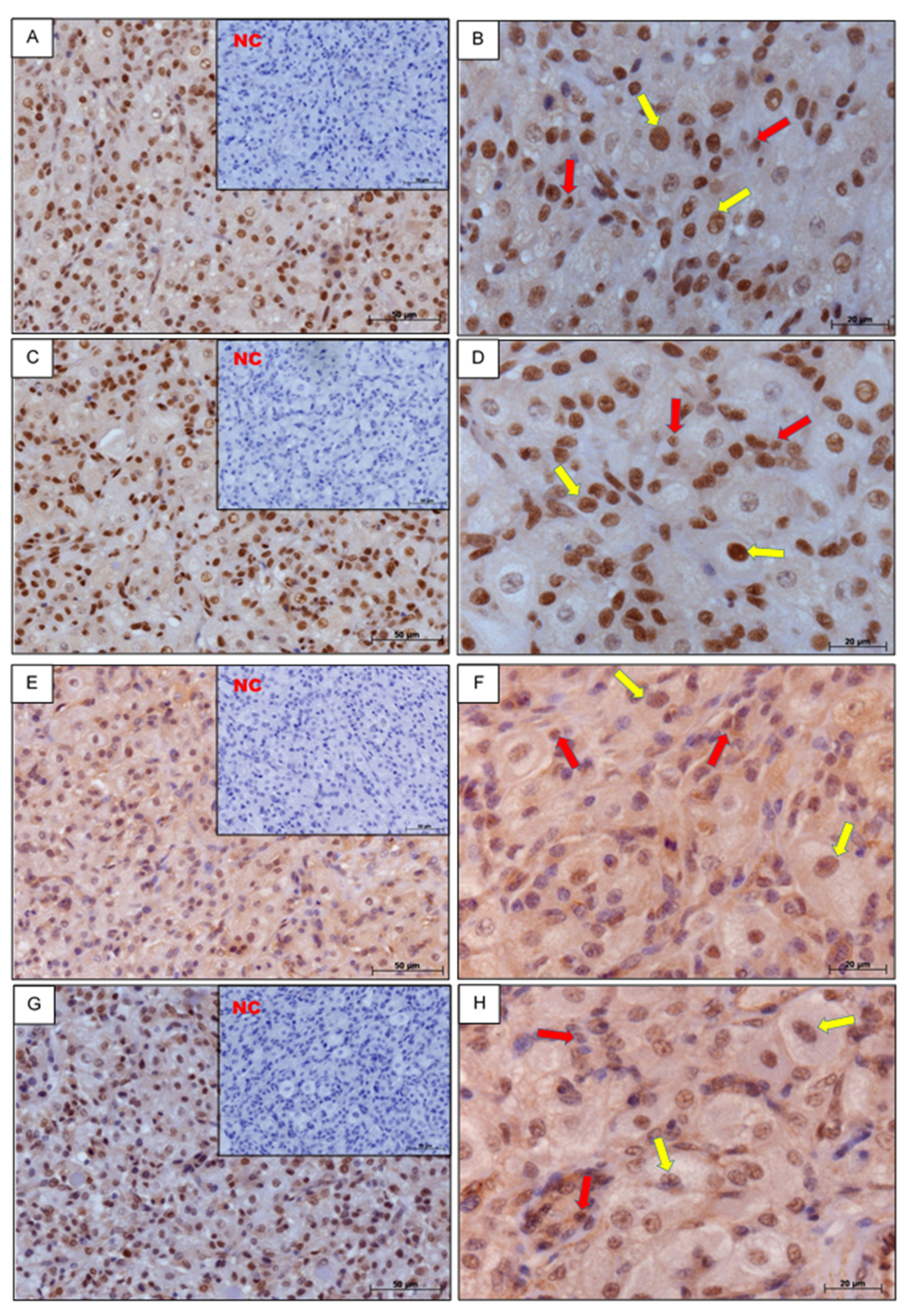
2.3. Immunohistochemistry
2.4. RNA Isolation and Reverse Transcription
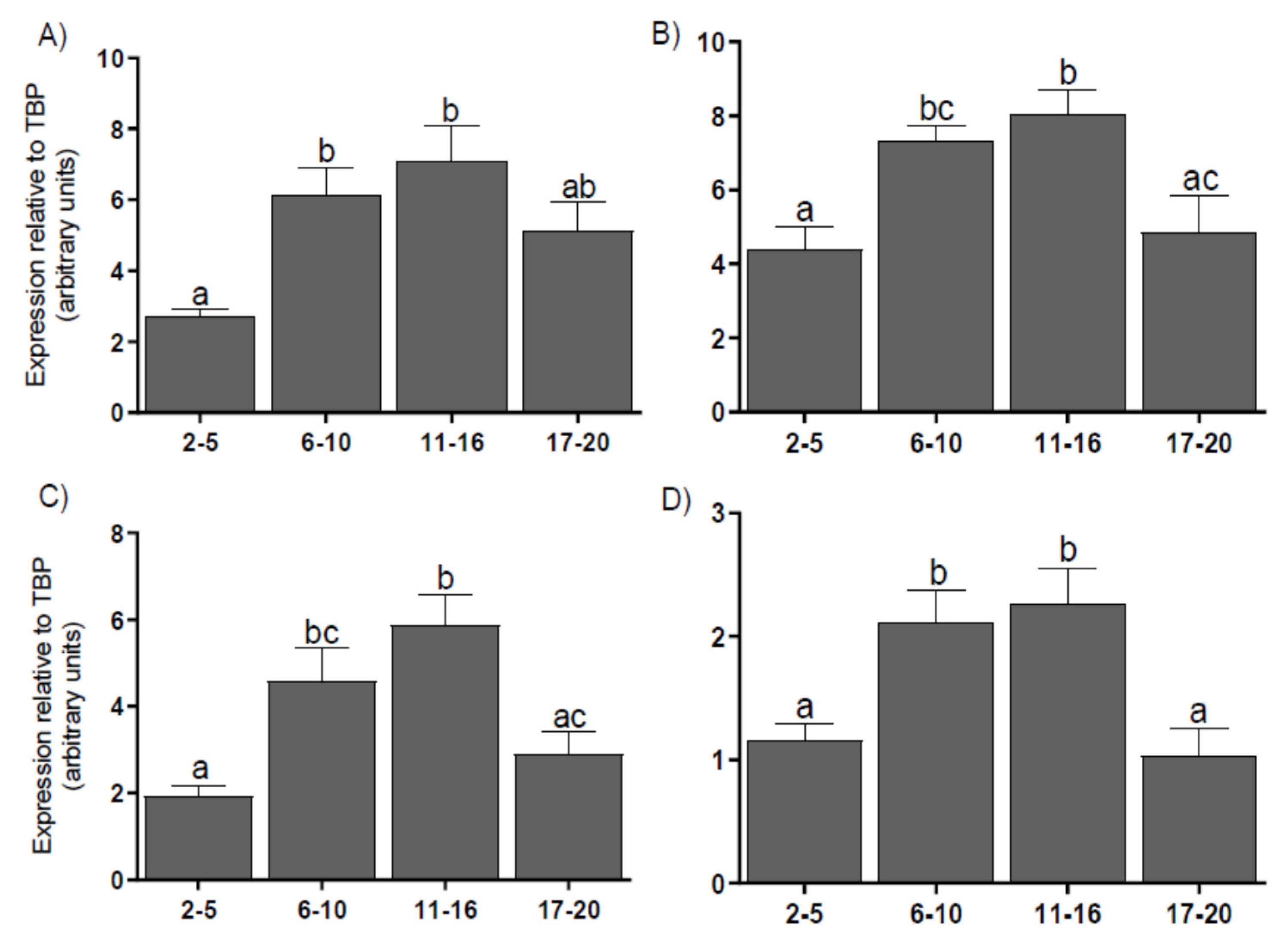
2.5. Real-Time PCR
2.6. Western Blot
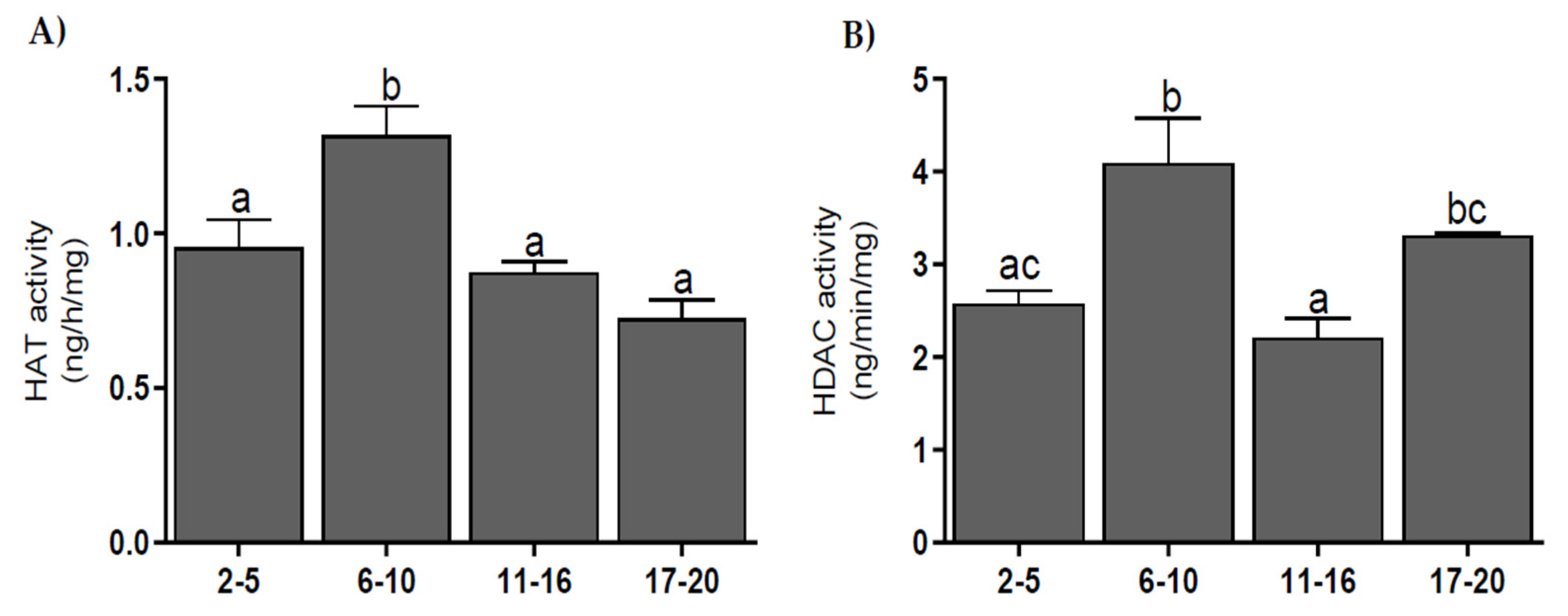
2.7. HAT and HDAC Activities
2.8. Data Analysis
3. Results
3.1. Immunohistochemistry
3.2. Progesterone Concentration
3.3. Expression of Coregulators mRNA in the CL
3.4. Protein Level of Coregulators in the CL
3.5. Histone Acetyltransferase and Histone Deacetylase Activities
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Niswender, G.D.; Juengel, J.L.; Silva, P.J.; Rollyson, M.K.; McIntush, E.W. Mechanisms controlling the function and life span of the corpus luteum. Physiol. Rev. 2000, 80, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Mulac-Jericevic, B.; Conneely, O.M. Reproductive tissue selective actions of progesterone receptors. Reproduction 2004, 128, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Pieber, D.; Allport, V.C.; Bennett, P.R. Progesterone receptor isoform A inhibits isoform B-mediated transactivation in human amnion. Eur. J. Pharmacol. 2001, 427, 7–11. [Google Scholar] [CrossRef]
- Cheung, J.; Smith, D.F. Molecular chaperone interactions with steroid receptors: An update. Mol. Endocrinol. 2000, 14, 939–946. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar]
- Xu, L.; Glass, C.K.; Rosenfeld, M.G. Coactivator and corepressor complexes in nuclear receptor function. Curr. Opin. Genet. Dev. 1999, 9, 140–147. [Google Scholar] [CrossRef]
- Tyler, J.K.; Kadonaga, J.T. The “dark side” of chromatin remodeling: Repressive effects on transcription. Cell 1999, 99, 443–446. [Google Scholar] [CrossRef]
- Lazar, M.A. Nuclear receptor corepressors. Nucl. Recept. Signal. 2003, 1, e001. [Google Scholar] [CrossRef]
- Shiozawa, T.; Shih, H.-C.; Miyamoto, T.; Feng, Y.-Z.; Uchikawa, J.; Itoh, K.; Konishi, I. Cyclic changes in the expression of steroid receptor coactivators and corepressors in the normal human endometrium. J. Clin. Endocrinol. Metab. 2003, 88, 871–878. [Google Scholar] [CrossRef][Green Version]
- Rekawiecki, R.; Kowalik, M.K.; Kotwica, J. The expression of progesterone receptor coregulators mRNA and protein in corpus luteum and endometrium of cows during the estrous cycle. Anim. Reprod. Sci. 2017, 183, 102–109. [Google Scholar] [CrossRef]
- Ireland, J.J.; Murphee, R.L.; Coulson, P.B. Accuracy of predicting stages of bovine estrous cycle by gross appearance of the corpus luteum. J. Dairy Sci. 1980, 63, 155–160. [Google Scholar] [CrossRef]
- Prakash, B.S.; Meyer, H.H.; Schallenberger, E.; van de Wiel, D.F. Development of a sensitive enzymeimmunoassay (EIA) for progesterone determination in unextracted bovine plasma using the second antibody technique. J. Steroid Biochem. 1987, 28, 623–627. [Google Scholar] [CrossRef]
- Tsang, P.C.; Walton, J.S.; Hansel, W. Oxytocin-specific RNA, oxytocin and progesterone concentrations in corpora lutea of heifers treated with oxytocin. J. Reprod. Fertil. 1990, 89, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kotwica, J.; Skarzynski, D.J.; Jaroszewski, J.J.; Bogacki, M. Noradrenaline affects secretory function of corpus luteum independently on prostaglandins in conscious cattle. Prostaglandins 1994, 48, 1–10. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A Defined Methodology for Reliable Quantification of Western Blot Data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef]
- Zhao, S.; Fernald, R.D. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 2005, 12, 1047–1064. [Google Scholar] [CrossRef]
- Sakumoto, R.; Vermehren, M.; Kenngott, R.A.-M.; Okuda, K.; Sinowatz, F. Changes in the levels of progesterone receptor mRNA and protein in the bovine corpus luteum during the estrous cycle. J. Reprod. Dev. 2010, 56, 219–222. [Google Scholar] [CrossRef]
- Rekawiecki, R.; Kowalik, M.K.; Kotwica, J. Cloning and expression of progesterone receptor isoforms A and B in bovine corpus luteum. Reprod. Fertil. Dev. 2014. [Google Scholar] [CrossRef]
- Dasgupta, S.; Lonard, D.M.; O’Malley, B.W. Nuclear receptor coactivators: Master regulators of human health and disease. Annu. Rev. Med. 2014, 65, 279–292. [Google Scholar] [CrossRef]
- Rowan, B.G.; O’Malley, B.W. Progesterone receptor coactivators. Steroids 2000, 65, 545–549. [Google Scholar] [CrossRef]
- Kotwica, J.; Rekawiecki, R.; Duras, M. Stimulatory influence of progesterone on its own synthesis in bovine corpus luteum. Bull.-Vet. Inst. Pulawy 2004, 48, 139–145. [Google Scholar]
- Rekawiecki, R.; Nowik, M.; Kotwica, J. Stimulatory effect of LH, PGE2 and progesterone on StAR protein, cytochrome P450 cholesterol side chain cleavage and 3beta hydroxysteroid dehydrogenase gene expression in bovine luteal cells. Prostaglandins Other Lipid Mediat. 2005, 78, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Rekawiecki, R.; Kotwica, J. Molecular regulation of progesterone (P4) synthesis within the bovine corpus luteum (CL). Vet. Med.-Czech 2007, 52, 405–412. [Google Scholar] [CrossRef]
- Rekawiecki, R.; Kowalik, M.K.; Slonina, D.; Kotwica, J. Regulation of progesterone synthesis and action in bovine corpus luteum. J. Physiol. Pharmacol. 2008, 59 (Suppl. 9), 75–89. [Google Scholar]
- Van den Broeck, W.; D’haeseleer, M.; Coryn, M.; Simoens, P. Cell-specific distribution of progesterone receptors in the bovine ovary. Reprod. Domest. Anim. 2002, 37, 314–320. [Google Scholar] [CrossRef]
- Salvetti, N.R.; Alfaro, N.S.; Velázquez, M.M.L.; Amweg, A.N.; Matiller, V.; Díaz, P.U.; Ortega, H.H. Alteration in localization of steroid hormone receptors and coregulatory proteins in follicles from cows with induced ovarian follicular cysts. Reproduction 2012, 144, 723–735. [Google Scholar] [CrossRef]
- Berisha, B.; Pfaffl, M.W.; Schams, D. Expression of estrogen and progesterone receptors in the bovine ovary during estrous cycle and pregnancy. Endocrine 2002, 17, 207–214. [Google Scholar] [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef]
- Kato, S.; Yokoyama, A.; Fujiki, R. Nuclear receptor coregulators merge transcriptional coregulation with epigenetic regulation. Trends Biochem. Sci. 2011, 36, 272–281. [Google Scholar] [CrossRef]
- Zecchin, A.; Pattarini, L.; Gutierrez, M.I.; Mano, M.; Mai, A.; Valente, S.; Myers, M.P.; Pantano, S.; Giacca, M. Reversible acetylation regulates vascular endothelial growth factor receptor-2 activity. J. Mol. Cell. Biol. 2014, 6, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Kovacs, M.; Chellappan, S. Regulation of vascular endothelial growth factor receptors by Rb and E2F1: Role of acetylation. Cancer Res. 2010, 70, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Kliem, H.; Berisha, B.; Meyer, H.H.D.; Schams, D. Regulatory changes of apoptotic factors in the bovine corpus luteum after induced luteolysis. Mol. Reprod. Dev. 2009, 76, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Heery, D.M.; Kalkhoven, E.; Hoare, S.; Parker, M.G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997, 387, 733–736. [Google Scholar] [CrossRef]
- Perissi, V.; Staszewski, L.M.; McInerney, E.M.; Kurokawa, R.; Krones, A.; Rose, D.W.; Lambert, M.H.; Milburn, M.V.; Glass, C.K.; Rosenfeld, M.G. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999, 13, 3198–3208. [Google Scholar] [CrossRef]
- Nagy, L.; Kao, H.Y.; Love, J.D.; Li, C.; Banayo, E.; Gooch, J.T.; Krishna, V.; Chatterjee, K.; Evans, R.M.; Schwabe, J.W. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999, 13, 3209–3216. [Google Scholar] [CrossRef]
- Tagami, T.; Gu, W.-X.; Peairs, P.T.; West, B.L.; Jameson, J.L. A Novel Natural Mutation in the Thyroid Hormone Receptor Defines a Dual Functional Domain That Exchanges Nuclear Receptor Corepressors and Coactivators. Mol. Endocrinol. 1998, 12, 1888–1902. [Google Scholar] [CrossRef]
- Peserico, A.; Simone, C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef]
- Icardi, L.; De Bosscher, K.; Tavernier, J. The HAT/HDAC interplay: Multilevel control of STAT signaling. Cytokine Growth Factor Rev. 2012, 23, 283–291. [Google Scholar] [CrossRef]
- Sandhoff, T.W.; McLean, M.P. Prostaglandin F2α reduces steroidogenic acute regulatory (StAR) protein messenger ribonucleic acid expression in the rat ovary. Endocrine 1996, 5, 183–190. [Google Scholar] [CrossRef]
- Liu, Q.; Merkler, K.A.; Zhang, X.; McLean, M.P. Prostaglandin F2alpha suppresses rat steroidogenic acute regulatory protein expression via induction of Yin Yang 1 protein and recruitment of histone deacetylase 1 protein. Endocrinology 2007, 148, 5209–5219. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen Name | Primers | GenBank Accession Number | Amplicon Length |
|---|---|---|---|
| P300 | F: CCATGAGCAACATGAGTGCTAGT R: CATTGTCACTCATCAGTGGGTTTT | XM_027540695.1 | 129 |
| CREB | F: TGAAGTGAAGGTCGAAGCTAAAGA R: GTACAGAGCTTCCAGGGTTGACAT | XM_024984694.1 | 147 |
| SRC-1 | F: CCCAGGCAGACGCTAAACAG R: TCAAGATAGCTTGCCGATTTTG | XM_028514416.1 | 114 |
| NCOR-2 | F: AGCCCTCGAGGCAAAAGC R: CATGCGGAGAGGCCTTGA | XM_024977670.1 | 177 |
| TBP | F: CAGAGAGCTCCGGGATCGT R: ACACCATCTTCCCAGAACTGAATAT | NM_001075742 | 194 |
| P300 | CREB | SRC-1 | NCOR-2 | |
|---|---|---|---|---|
| P4–mRNA | r = 0.59 p < 0.001 | r = 0.56 p < 0.05 | r = 0.76 p < 0.0001 | r = 0.79 p < 0.0001 |
| P4–protein | ns | ns | ns | ns |
| mRNA corepressor mRNA coactivators | r = 0.79 p < 0.0001 | r = 0.75 p < 0.0001 | r = 0.89 p < 0.0001 | − |
| Protein corepressor Protein coactivator | ns | r = 0.79 p < 0.01 | ns | − |
| HAT–HDAC | r = 0.51 p < 0.05 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rekawiecki, R.; Dobrzyn, K.; Kotwica, J.; Kowalik, M.K. Progesterone Receptor Coregulators as Factors Supporting the Function of the Corpus Luteum in Cows. Genes 2020, 11, 923. https://doi.org/10.3390/genes11080923
Rekawiecki R, Dobrzyn K, Kotwica J, Kowalik MK. Progesterone Receptor Coregulators as Factors Supporting the Function of the Corpus Luteum in Cows. Genes. 2020; 11(8):923. https://doi.org/10.3390/genes11080923
Chicago/Turabian StyleRekawiecki, Robert, Karolina Dobrzyn, Jan Kotwica, and Magdalena K. Kowalik. 2020. "Progesterone Receptor Coregulators as Factors Supporting the Function of the Corpus Luteum in Cows" Genes 11, no. 8: 923. https://doi.org/10.3390/genes11080923
APA StyleRekawiecki, R., Dobrzyn, K., Kotwica, J., & Kowalik, M. K. (2020). Progesterone Receptor Coregulators as Factors Supporting the Function of the Corpus Luteum in Cows. Genes, 11(8), 923. https://doi.org/10.3390/genes11080923