The STRidER Report on Two Years of Quality Control of Autosomal STR Population Datasets



Abstract
1. Introduction
2. Autosomal STR Data Quality Control on STRidER
2.1. What STRidER Quality Control Is (Not): Rationale and Workflow
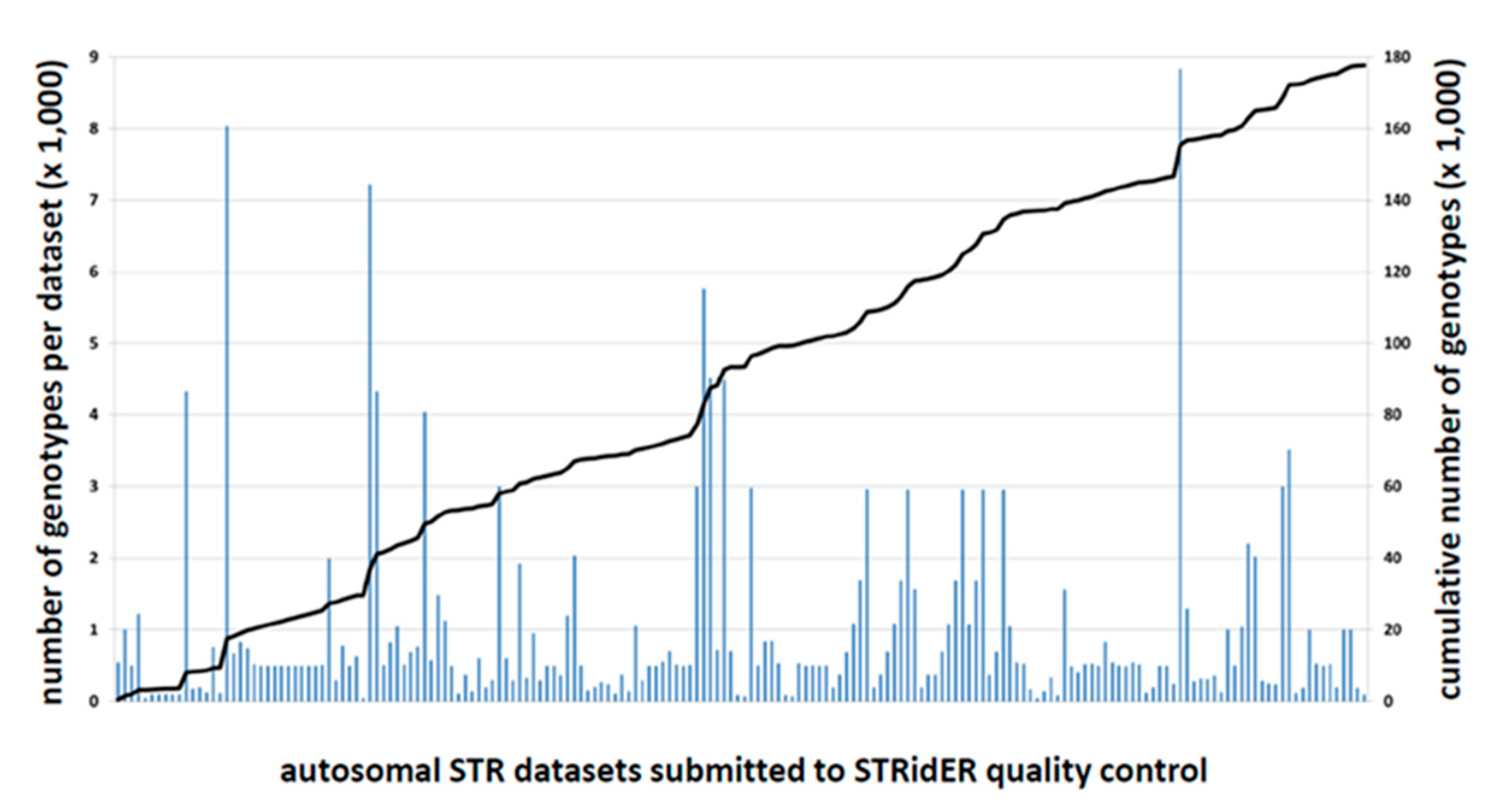
2.2. STRidER Quality Control Submissions in the First Two Years
3. Results of STRidER Quality Control
3.1. Overall Outcome
3.2. Error Statistics
3.2.1. CE Dataset Error Charts
3.2.2. MPS Dataset Errors
4. Discussion
4.1. Origin and Effect of Errors in Autosomal STR Datasets
4.2. Literature Reports on Errors in Forensic Autosomal STR Data
4.3. The Evolution of STRidER Quality Control in Its First Two Years
4.4. Reactions on STRidER Quality Control
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jobling, M.A.; Gill, P. Encoded evidence: DNA in forensic analysis. Nat. Rev. Genet. 2004, 5, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.M. Scientific standards for studies in forensic genetics. Forensic Sci. Int. 2007, 165, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; Bottrell, M.C.; Bunch, S.G.; Fram, R.; Harrison, D.; Meagher, S.; Oien, C.T.; Peterson, P.E.; Seiger, D.P.; Smith, M.B.; et al. A perspective on errors, bias, and interpretation in the forensic sciences and direction for continuing advancement. J. Forensic Sci. 2009, 54, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Coble, M.D.; Kline, M.C.; Butler, J.M. Metrology needs and NIST resources for the forensic DNA community. Accredit. Qual. Assur. 2011, 16, 293–297. [Google Scholar] [CrossRef]
- Ansell, R. Internal quality control in forensic DNA analysis. Accredit. Qual. Assur. 2013, 18, 279–289. [Google Scholar] [CrossRef]
- Ricci, U. Establishment of an ISO 17025:2005 accredited forensic genetics laboratory in Italy. Accredit. Qual. Assur. 2014, 19, 289–299. [Google Scholar] [CrossRef]
- Butler, J.M. U.S. Initiatives to strengthen forensic science & international standards in forensic DNA. Forensic Sci. Int. Genet. 2015, 18, 4–20. [Google Scholar]
- ENFSI DNA Working Group. DNA Database Management—Review and Recommendations; ENFSI: Wiesbaden, Germany, 2017. [Google Scholar]
- National Research Council. Strengthening Forensic Science in the US: A Path Forward; The National Academies Press: Washington, DC, USA, 2009. [Google Scholar]
- SWGDAM Quality Assurance Standards for Forensic DNA Testing Laboratories and for DNA Databasing Laboratories (effective 1 July 2020). Available online: https://www.swgdam.org/publications (accessed on 5 August 2020).
- Butler, J.M.; Willis, S. Interpol review of forensic biology and forensic DNA typing 2016–2019. Forensic Sci. Int. Synerg. 2020. [Google Scholar] [CrossRef]
- Parson, W.; Dür, A. EMPOP—A forensic mtDNA database. Forensic Sci. Int. Genet. 2007, 1, 88–92. [Google Scholar] [CrossRef]
- Willuweit, S.; Roewer, L. The new Y chromosome haplotype reference database. Forensic Sci. Int. Genet. 2015, 15, 43–48. [Google Scholar] [CrossRef]
- Bodner, M.; Bastisch, I.; Butler, J.M.; Fimmers, R.; Gill, P.; Gusmao, L.; Morling, N.; Phillips, C.; Prinz, M.; Schneider, P.M.; et al. Recommendations of the DNA commission of the International Society for Forensic Genetics (ISFG) on quality control of autosomal short tandem repeat allele frequency databasing (STRidER). Forensic Sci. Int. Genet. 2016, 24, 97–102. [Google Scholar] [CrossRef]
- Gouy, A.; Zieger, M. STRAF—a convenient online tool for STR data evaluation in forensic genetics. Forensic Sci. Int. Genet. 2017, 30, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Brandstätter, A.; Alonso, A.; Brandt, N.; Brinkmann, B.; Carracedo, A.; Corach, D.; Froment, O.; Furac, I.; Grzybowski, T.; et al. The EDNAP mitochondrial DNA population database (EMPOP) collaborative exercises: Organisation, results and perspectives. Forensic Sci. Int. 2004, 139, 215–226. [Google Scholar] [CrossRef]
- Gusmao, L.; Butler, J.M.; Linacre, A.; Parson, W.; Roewer, L.; Schneider, P.M.; Carracedo, A. Revised guidelines for the publication of genetic population data. Forensic Sci. Int. Genet. 2017, 30, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Roewer, L. Publication of population data of linearly inherited DNA markers in the International Journal of Legal Medicine. Int. J. Leg. Med. 2010, 124, 505–509. [Google Scholar] [CrossRef]
- Poetsch, M.; Bajanowski, T.; Pfeiffer, H. The publication of population genetic data in the International Journal of Legal Medicine: Guidelines. Int. J. Leg. Med. 2012, 126, 489–490. [Google Scholar] [CrossRef][Green Version]
- Martin, P.D.; Schmitter, H.; Schneider, P.M. A brief history of the formation of DNA databases in forensic science within Europe. Forensic Sci. Int. 2001, 119, 225–231. [Google Scholar] [CrossRef]
- Presciuttini, S.; Cerri, N.; Turrina, S.; Pennato, B.; Alu, M.; Asmundo, A.; Barbaro, A.; Boschi, I.; Buscemi, L.; Caenazzo, L.; et al. Validation of a large Italian database of 15 STR loci. Forensic Sci. Int. 2006, 156, 266–268. [Google Scholar] [CrossRef]
- Welch, L.A.; Gill, P.; Phillips, C.; Ansell, R.; Morling, N.; Parson, W.; Palo, J.U.; Bastisch, I. European Network of Forensic Science Institutes (ENFSI): Evaluation of new commercial STR multiplexes that include the European standard set (ESS) of markers. Forensic Sci. Int. Genet. 2012, 6, 819–826. [Google Scholar] [CrossRef]
- Pemberton, T.J.; DeGiorgio, M.; Rosenberg, N.A. Population structure in a comprehensive genomic data set on human microsatellite variation. G3 Genes Genom. Genet. 2013, 3, 891–907. [Google Scholar] [CrossRef]
- Gomes, S.M.; Bodner, M.; Souto, L.; Zimmermann, B.; Huber, G.; Strobl, C.; Röck, A.W.; Achilli, A.; Olivieri, A.; Torroni, A.; et al. Human settlement history between sunda and sahul: A focus on East Timor (Timor-Leste) and the Pleistocenic mtDNA diversity. BMC Genom. 2015, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Moretti, T.R.; Budowle, B.; Buckleton, J.S. Erratum. J. Forensic Sci. 2015, 60, 1114–1116. [Google Scholar] [CrossRef] [PubMed]
- Buckleton, J.; Curran, J.; Goudet, J.; Taylor, D.; Thiery, A.; Weir, B.S. Population-specific Fst values for forensic STR markers: A worldwide survey. Forensic Sci. Int. Genet. 2016, 23, 91–100. [Google Scholar] [CrossRef]
- Gill, P.; Foreman, L.; Buckleton, J.S.; Triggs, C.M.; Allen, H. A comparison of adjustment methods to test the robustness of an STR DNA database comprised of 24 European populations. Forensic Sci. Int. 2003, 131, 184–196. [Google Scholar] [CrossRef]
- Previderè, C.; Grignani, P.; Presciuttini, S. Italian population data for the new ENFSI/EDNAP loci D1S1656, D2S441, D10S1248, D12S391, D22S1045. The GeFi collaborative exercise and concordance study. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e238–e239. [Google Scholar] [CrossRef]
- Parson, W.; Ballard, D.; Budowle, B.; Butler, J.M.; Gettings, K.B.; Gill, P.; Gusmao, L.; Hares, D.R.; Irwin, J.A.; King, J.L.; et al. Massively parallel sequencing of forensic STRs: Considerations of the DNA commission of the International Society for Forensic Genetics (ISFG) on minimal nomenclature requirements. Forensic Sci. Int. Genet. 2016, 22, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R. Sample size requirements for addressing the population genetic issues of forensic use of DNA typing. Hum. Biol. 1992, 64, 141–159. [Google Scholar]
- Restrepo, T.; Martinez, M.; Palacio, O.; Posada, Y.; Zapata, S.; Gusmão, L.; Ibarra, A. Database sample size effect on minimum allele frequency estimation: Database comparison analysis of samples of 4652 and 560 individuals for 22 microsatellites in Colombian population. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e13–e14. [Google Scholar] [CrossRef]
- National Research Council. The Evaluation of Forensic DNA Evidence; The National Academies Press: Washington, DC, USA, 1996. [Google Scholar]
- Rand, S.; Schurenkamp, M.; Hohoff, C.; Brinkmann, B. The GEDNAP blind trial concept part II. Trends and developments. Int. J. Leg. Med. 2004, 118, 83–89. [Google Scholar] [CrossRef]
- Phillips, C. A genomic audit of newly-adopted autosomal STRs for forensic identification. Forensic Sci. Int. Genet. 2017, 29, 193–204. [Google Scholar] [CrossRef]
- Novroski, N.M.M.; Wendt, F.R.; Woerner, A.E.; Bus, M.M.; Coble, M.; Budowle, B. Expanding beyond the current core STR loci: An exploration of 73 STR markers with increased diversity for enhanced DNA mixture deconvolution. Forensic Sci. Int. Genet. 2019, 38, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Parson, W.; Amigo, J.; King, J.L.; Coble, M.D.; Steffen, C.R.; Vallone, P.M.; Gettings, K.B.; Butler, J.M.; Budowle, B. D5S2500 is an ambiguously characterized STR: Identification and description of forensic microsatellites in the genomics age. Forensic Sci. Int. Genet. 2016, 23, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Gettings, K.B.; Borsuk, L.A.; Ballard, D.; Bodner, M.; Budowle, B.; Devesse, L.; King, J.; Parson, W.; Phillips, C.; Vallone, P.M. STRSeq: A catalog of sequence diversity at human identification short tandem repeat loci. Forensic Sci. Int. Genet. 2017, 31, 111–117. [Google Scholar] [CrossRef]
- Phillips, C.; Gettings, K.B.; King, J.L.; Ballard, D.; Bodner, M.; Borsuk, L.; Parson, W. “The devil’s in the detail”: Release of an expanded, enhanced and dynamically revised forensic STR sequence guide. Forensic Sci. Int. Genet. 2018, 34, 162–169. [Google Scholar] [CrossRef]
- Gettings, K.B.; Ballard, D.; Bodner, M.; Borsuk, L.A.; King, J.L.; Parson, W.; Phillips, C. Report from the STRAND working group on the 2019 STR sequence nomenclature meeting. Forensic Sci. Int. Genet. 2019, 43, 102165. [Google Scholar] [CrossRef]
- Alonso, A.; Müller, P.; Roewer, L.; Willuweit, S.; Budowle, B.; Parson, W. European survey on forensic applications of massively parallel sequencing. Forensic Sci. Int. Genet. 2017, 29, e23–e25. [Google Scholar] [CrossRef]
- Lincoln, P.; Carracedo, A. Publication of population data of human polymorphisms. Forensic Sci. Int. Genet. 2000, 110, 3–5. [Google Scholar]
- Evett, I.W. Trivial error. Nature 1991, 354, 114. [Google Scholar] [CrossRef]
- Curnow, R.N. Discussion of Berry, D.A.; Evett, I.W.; Pinchin, R. Statistical inference in crime investigations using deoxyribonucleic acid profiling. J. R. Stat. Soc. Ser. C 1992, 41, 521–522. [Google Scholar]
- Steffen, C.R.; Coble, M.D.; Gettings, K.B.; Vallone, P.M. Corrigendum to ‘U.S. Population data for 29 autosomal STR loci’ [Forensic Sci. Int. Genet. 7 (2013) e82–e83]. Forensic Sci. Int. Genet. 2017, 31, e36–e40. [Google Scholar] [CrossRef]
- Brenner, C.H. Fundamental problem of forensic mathematics-the evidential value of a rare haplotype. Forensic Sci. Int. Genet. 2010, 4, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, A.R.; Hallenberg, C.; Simonsen, B.T.; Langkjaer, R.B.; Morling, N. A report of the 2002–2008 paternity testing workshops of the English speaking working group of the International Society for Forensic Genetics. Forensic Sci. Int. Genet. 2009, 3, 214–221. [Google Scholar] [CrossRef]
- Purser, M.; Chadwick, D. Does an awareness of differing types of spreadsheet errors aid end-users in identifying spreadsheets errors? arXiv 2006, arXiv:0803.0167. [Google Scholar]
- Rand, S.; Schurenkamp, M.; Brinkmann, B. The GEDNAP (German DNA profiling group) blind trial concept. Int. J. Leg. Med. 2002, 116, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, B.; Butler, R.; Lincoln, P.; Mayr, W.R.; Rossi, U.; Bär, W.; Baur, M.; Budowle, B.; Fimmers, R.; Gill, P.; et al. 1991 report concerning recommendations of the DNA commission of the International Society for Forensic Haemogenetics relating to the use of DNA polymorphisms. Forensic Sci. Int. 1992, 52, 125–130. [Google Scholar]
- National Research Council. DNA Technology in Forensic Science; The National Academies Press: Washington, DC, USA, 1992. [Google Scholar]
- Budowle, B.; Monson, K.L.; Giusti, A.M.; Brown, B.L. The assessment of frequency estimates of HaeIII-generated VNTR profiles in various reference databases. J. Forensic Sci. 1994, 39, 319–352. [Google Scholar]
- Budowle, B.; Monson, K.L.; Chakraborty, R. Estimating minimum allele frequencies for DNA profile frequency estimates for PCR-based loci. Int. J. Leg. Med. 1996, 108, 173–176. [Google Scholar] [CrossRef]
- Pamplona, J.P.; Freitas, F.; Pereira, L. A worldwide database of autosomal markers used by the forensic community. Forensic Sci. Int. Genet. Suppl. Ser. 2008, 1, 656–657. [Google Scholar] [CrossRef]
- Wilson-Wilde, L.; Smith, S.; Bruenisholz, E. The analysis of Australian proficiency test data over a ten-year period. Forensic Sci. Policy Manag. Int. J. 2017, 8, 55–63. [Google Scholar] [CrossRef]
- Gill, P.; Kimpton, C.; D’Aloja, E.; Andersen, J.F.; Bär, W.; Brinkmann, B.; Holgersson, S.; Johnsson, V.; Kloosterman, A.D.; Lareu, M.V.; et al. Report of the European DNA profiling group (EDNAP)-towards standardisation of short tandem repeat (STR) loci. Forensic Sci. Int. 1994, 65, 51–59. [Google Scholar] [CrossRef]
- Fernández, K.; Gómez, J.; García-Hirschfeld, J.; Cubillo, E.; De La Torre, C.S.; Vallejo, G. Accreditation of the GHEP-ISFG proficiency test: One step forward to assure and improve quality. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e515–e517. [Google Scholar] [CrossRef]
- Peterson, J.L.; Lin, G.; Ho, M.; Chen, Y.; Gaensslen, R.E. The feasibility of external blind DNA proficiency testing. I. Background and findings. J. Forensic Sci. 2003, 48, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Koehler, J.J. Proficiency tests to estimate error rates in the forensic sciences. Law Probab. Risk 2012, 12, 89–98. [Google Scholar] [CrossRef]
- Butler, J.M.; Kline, M.C.; Coble, M.D. NIST interlaboratory studies involving DNA mixtures (MIX05 and MIX13): Variation observed and lessons learned. Forensic Sci. Int. Genet. 2018, 37, 81–94. [Google Scholar] [CrossRef]
- Kloosterman, A.; Sjerps, M.; Quak, A. Error rates in forensic DNA analysis: Definition, numbers, impact and communication. Forensic Sci. Int. Genet. 2014, 12, 77–85. [Google Scholar] [CrossRef]
- Gomez, J.; Carracedo, A. The 1998–1999 collaborative exercises and proficiency testing program on DNA typing of the Spanish and Portuguese Working Group of the International Society for Forensic Genetics (GEP-ISFG). Forensic Sci. Int. 2000, 114, 21–30. [Google Scholar] [CrossRef]
- Hallenberg, C.; Morling, N. A report of the 1997, 1998 and 1999 paternity testing workshops of the English Speaking Working Group of the International Society for Forensic Genetics. Forensic Sci. Int. 2001, 116, 23–33. [Google Scholar] [CrossRef]
- Hallenberg, C.; Morling, N. A report of the 2000 and 2001 paternity testing workshops of the English Speaking Working Group of the International Society for Forensic Genetics. Forensic Sci. Int. 2002, 129, 43–50. [Google Scholar] [CrossRef]
- Gómez, J.; Alonso, A.; García, O.; Carracedo, A. The proficiency testing program on DNA typing of the Spanish and Portuguese working group of the ISFG. Int. Congr. Ser. 2003, 1239, 837–840. [Google Scholar] [CrossRef]
- Gómez, J.; García-Hirschfeld, J.; García, O.; Carracedo, A. GEP proficiency testing program in forensic genetics: 10 years of experience. Int. Congr. Ser. 2004, 1261, 124–126. [Google Scholar] [CrossRef]
- García-Hirschfeld, J.; Alonso, A.; García, O.; Amorim, A.; Gómez, J. 2004–2005 GEP proficiency testing programs: Special emphasis on the interlaboratory analysis of mixed stains. Int. Congr. Ser. 2006, 1288, 855–857. [Google Scholar] [CrossRef]
- Hallenberg, C.; Langkjær, R.B.; Jensen, P.B.; Simonsen, B.T.; Morling, N. Results of the 2007 paternity testing workshop of English Speaking Working Group of the International Society for Forensic Genetics. Forensic Sci. Int. Genet. Suppl. Ser. 2008, 1, 680–681. [Google Scholar] [CrossRef]
- García-Hirschfeld, J.; Alonso, A.; García, O.; Amorim, A.; Gusmão, L.; Luque, J.A.; van Asch, B.; Carracedo, A.; Gómez, J. GEP-ISFG proficiency testing programs: 2007 update. Forensic Sci. Int. Genet. Suppl. Ser. 2008, 1, 674–676. [Google Scholar] [CrossRef]
- Friis, S.L.; Hallenberg, C.; Simonsen, B.T.; Morling, N. Results of the 2009 paternity testing workshop of the English Speaking Working Group of the International Society for Forensic Genetics. Forensic Sci. Int. Genet. Suppl. Ser. 2009, 2, 91–92. [Google Scholar] [CrossRef]
- Poulsen, L.; Friis, S.L.; Hallenberg, C.; Simonsen, B.T.; Morling, N. Results of the 2011 relationship testing workshop of the English Speaking Working Group. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e512–e513. [Google Scholar] [CrossRef]
- Friis, S.L.; Hallenberg, C.; Simonsen, B.T.; Morling, N. Results of the 2013 relationship testing workshop of the English Speaking Working Group. Forensic Sci. Int. Genet. Suppl. Ser. 2013, 4, e282–e283. [Google Scholar] [CrossRef]
- Friis, S.L.; Hallenberg, C.; Simonsen, B.T.; Morling, N. Results of the 2015 relationship testing workshop of the English Speaking Working Group. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e320–e321. [Google Scholar] [CrossRef]
- Wilson-Wilde, L.; Romano, H.; Smith, S. Error rates in proficiency testing in Australia. Aust. J. Forensic Sci. 2019, 51, S268–S271. [Google Scholar] [CrossRef]
- Crespillo, M.; Barrio, P.A.; Luque, J.A.; Alves, C.; Aler, M.; Alessandrini, F.; Andrade, L.; Barretto, R.M.; Bofarull, A.; Costa, S.; et al. GHEP-ISFG collaborative exercise on mixture profiles of autosomal STRs (GHEP-MIX01, GHEP-MIX02 and GHEP-MIX03): Results and evaluation. Forensic Sci. Int. Genet 2014, 10, 64–72. [Google Scholar] [CrossRef]
- Toscanini, U.; Gusmao, L.; Alava Narvaez, M.C.; Alvarez, J.C.; Baldassarri, L.; Barbaro, A.; Berardi, G.; Betancor Hernandez, E.; Camargo, M.; Carreras-Carbonell, J.; et al. Analysis of uni and bi-parental markers in mixture samples: Lessons from the 22nd GHEP-ISFG intercomparison exercise. Forensic Sci. Int. Genet. 2016, 25, 63–72. [Google Scholar] [CrossRef]
- Gomez, J.; Rodriguez-Calvo, M.S.; Albarran, C.; Amorim, A.; Andradas, J.; Cabrero, C.; Calvet, R.; Corach, D.; Crespillo, M.; Doutremepuich, C.; et al. A review of the collaborative exercises on DNA typing of the Spanish and Portuguese ISFH working group. International Society for Forensic Haemogenetics. Int. J. Leg. Med. 1997, 110, 273–277. [Google Scholar]
- Previdere, C.; Grignani, P.; Alessandrini, F.; Alu, M.; Biondo, R.; Boschi, I.; Caenazzo, L.; Carboni, I.; Carnevali, E.; De Stefano, F.; et al. The 2011 GeFi collaborative exercise. Concordance study, proficiency testing and Italian population data on the new ENFSI/EDNAP loci D1S1656, D2S441, D10S1248, D12S391, D22S1045. Forensic Sci. Int. Genet. 2013, 7, e15–e18. [Google Scholar] [CrossRef] [PubMed]
- Presciuttini, S.; Ciampini, F.; Alu, M.; Cerri, N.; Dobosz, M.; Domenici, R.; Peloso, G.; Pelotti, S.; Piccinini, A.; Ponzano, E.; et al. Allele sharing in first-degree and unrelated pairs of individuals in the GeFi AmpflSTR profiler plus database. Forensic Sci. Int. 2003, 131, 85–89. [Google Scholar] [CrossRef]
- Budowle, B.; Moretti, T.R.; Baumstark, A.L.; Defenbaugh, D.A.; Keys, K.M. Population data on the thirteen codis core short tandem repeat loci in African Americans, U.S. Caucasians, Hispanics, Bahamians, Jamaicans, and Trinidadians. J. Forensic Sci. 1999, 44, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.R.; Duewer, D.L.; Kline, M.C.; Coble, M.D.; Butler, J.M. U.S. Population data for 29 autosomal STR loci. Forensic Sci. Int. Genet. 2013, 7, e82–e83. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Schoske, R.; Vallone, P.M.; Redman, J.W.; Kline, M.C. Allele frequencies for 15 autosomal STR loci on U.S. Caucasian, African American, and Hispanic populations. J. Forensic Sci. 2003, 48, 908–911. [Google Scholar] [CrossRef]
- Prieto, L.; Haned, H.; Mosquera, A.; Crespillo, M.; Aleman, M.; Aler, M.; Alvarez, F.; Baeza-Richer, C.; Dominguez, A.; Doutremepuich, C.; et al. EUROFORGEN-NoE collaborative exercise on LRmix to demonstrate standardization of the interpretation of complex DNA profiles. Forensic Sci. Int. Genet. 2014, 9, 47–54. [Google Scholar] [CrossRef]
- Weirich, V. Completely automated interpretation of reference samples. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 68–69. [Google Scholar] [CrossRef]
- Ferragut, J.F.; Pinto, N.; Amorim, A.; Picornell, A. Improving publication quality and the importance of post publication peer review: The illustrating example of X chromosome analysis and calculation of forensic parameters. Forensic Sci. Int. Genet. 2019, 38, e5–e7. [Google Scholar] [CrossRef]


{kind=link}
| All | CE 1 | MPS 2 | |
|---|---|---|---|
| Number of Genotypes | |||
| Total | 177,595 | 173,709 | 3886 |
| Mean per dataset | 965 | 1053 | 205 |
| Median per dataset | 506 | 522 | 140 |
| Number of Datasets | |||
| Total | 184 | 165 | 19 |
| Passed QC | 48 | 35 | 13 |
| Withdrawal during QC | 41 | 36 | 5 |
| Rejection by STRidER | 58 | 58 | 0 |
| QC pending | 37 | 36 | 1 |
| Proportions of Datasets with completed QC (%) | |||
| Acceptance rate | 32.7 | 27.1 | 72.2 |
| Withdrawal/Rejection rate | 67.3 | 72.9 | 27.8 |
| n | (%) | |||
|---|---|---|---|---|
| Datasets Revealing Errors | 158 | 95.8 | ||
| Error categories | (i) | Identical genotypes | 63 | 38.2 |
| (ii) | Non-ascending allele pairs | 58 | 35.2 | |
| (iii) | Allele nomenclature errors | 29 | 17.6 | |
| (iv) | Allele calling errors | 17 | 10.3 | |
| (v) | Incomplete genotypes | 16 | 9.7 | |
| (vi) | Errors in locus nomenclature | 10 | 6.1 | |
| (vii) | Aneuploidy | 9 | 5.5 | |
| (viii) | No raw data/shuffled data | 9 | 5.5 | |
| (ix) | Identical identifiers | 7 | 4.2 | |
| (x) | Information mismatch | 5 | 3.0 | |
| (xi) | Locus swapping | 3 | 1.8 | |
| (xii) | Loss of intermediate alleles | 2 | 1.2 | |
| Datasets Revealing No Errors | 7 | 4.2 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodner, M.; Parson, W. The STRidER Report on Two Years of Quality Control of Autosomal STR Population Datasets. Genes 2020, 11, 901. https://doi.org/10.3390/genes11080901
Bodner M, Parson W. The STRidER Report on Two Years of Quality Control of Autosomal STR Population Datasets. Genes. 2020; 11(8):901. https://doi.org/10.3390/genes11080901
Chicago/Turabian StyleBodner, Martin, and Walther Parson. 2020. "The STRidER Report on Two Years of Quality Control of Autosomal STR Population Datasets" Genes 11, no. 8: 901. https://doi.org/10.3390/genes11080901
APA StyleBodner, M., & Parson, W. (2020). The STRidER Report on Two Years of Quality Control of Autosomal STR Population Datasets. Genes, 11(8), 901. https://doi.org/10.3390/genes11080901