Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System

 , , , , ,
, , , , , 
 on behalf of the VISAGE Consortium
on behalf of the VISAGE Consortium
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Experimental Design
2.2. VISAGE Basic Tool for Predicting Appearance and Ancestry Using PowerSeq Chemistry
2.3. Library Preparation and Sequencing
2.4. Data Processing
3. Results and Discussion
3.1. Negative Control
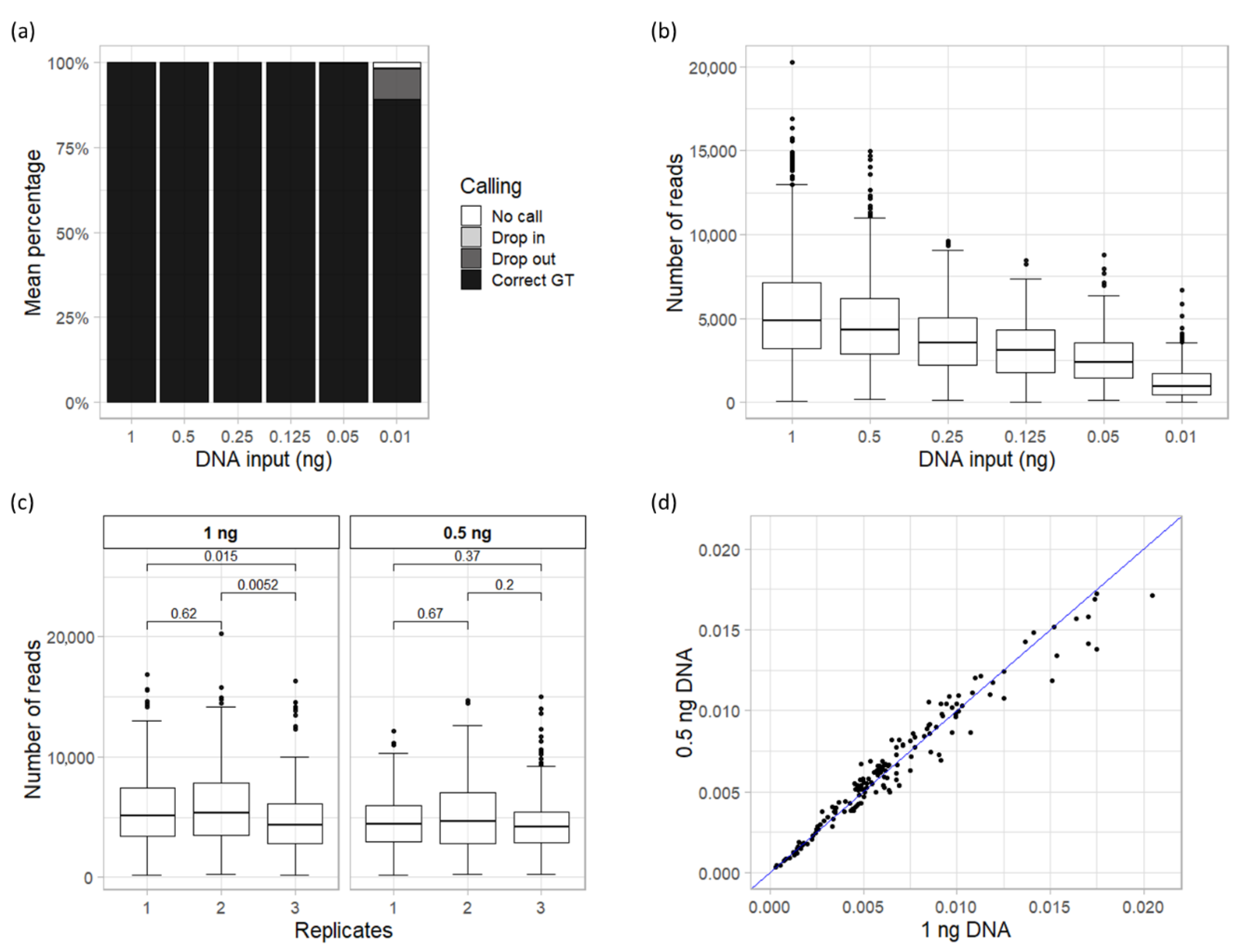
3.2. Sensitivity
3.3. Repeatability
3.4. Locus, Allele Balance and Base Misincorporation Rate
3.5. Casework “Mock” Samples
3.6. Concordance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
- Erasmus University Medical Center Rotterdam, Rotterdam, The Netherlands: Manfred Kayser, Vivian Kalamara, Arwin Ralf, Athina Vidaki
- Jagiellonian University, Krakow, Poland: Wojciech Branicki, Ewelina Pośpiech, Aleksandra Pisarek
- Universidade de Santiago de Compostela, Santiago de Compostela, Spain : Ángel Carracedo, Maria Victoria Lareu, Christopher Phillips, Ana Freire-Aradas, Ana Mosquera-Miguel, María de la Puente
- Medizinische Universität Innsbruck, Innsbruck, Austria: Walther Parson, Catarina Xavier, Antonia Heidegger, Harald Niederstätter
- Universität zu Köln, Cologne, Germany: Michael Nothnagel, Maria-Alexandra Katsara, Tarek Khellaf
- King’s College London, London, UK: Barbara Prainsack, Gabrielle Samuel
- Klinikum der Universität zu Köln, Cologne, Germany: Peter M. Schneider, Theresa E. Gross, Jan Fleckhaus
- Bundeskriminalamt, Wiesbaden, Germany: Ingo Bastisch, Nathalie Schury, Jens Teodoridis, Martina Unterländer
- Institut National De Police Scientifique, Lyon, France: François-Xavier Laurent, Caroline Bouakaze, Yann Chantrel, Anna Delest, Clémence Hollard, Ayhan Ulus, Julien Vannier
- Netherlands Forensic Institute, The Hague, Netherlands: Titia Sijen, Kris van der Gaag, Marina Ventayol-Garcia
- National Forensic Centre, Swedish Police Authority, Linköping, Sweden: Johannes Hedman, Klara Junker, Maja Sidstedt
- Metropolitan Police Service, London, United Kingdom: Shazia Khan, Carole E. Ames, Andrew Revoir
- Centralne Laboratorium Kryminalistyczne Policji, Warsaw, Poland: Magdalena Spólnicka, Ewa Kartasinska, Anna Woźniak
References
- Kayser, M. Forensic DNA Phenotyping: Predicting human appearance from crime scene material for investigative purposes. Forensic Sci. Int. Genet. 2015, 18, 33–48. [Google Scholar] [CrossRef]
- Phillips, C. Forensic genetic analysis of bio-geographical ancestry. Forensic Sci. Int. Genet. 2015, 18, 49–65. [Google Scholar] [CrossRef]
- Schneider, P.M.; Prainsack, B.; Kayser, M. The Use of Forensic DNA Phenotyping in Predicting Appearance and Biogeographic Ancestry. Dtsch. Arztebl. Int. 2019, 116, 873–880. [Google Scholar] [CrossRef]
- Butler, K.; Peck, M.; Hart, J.; Schanfield, M.; Podini, D. Molecular “eyewitness”: Forensic prediction of phenotype and ancestry. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e498–e499. [Google Scholar] [CrossRef]
- Walsh, S.; Chaitanya, L.; Breslin, K.; Muralidharan, C.; Bronikowska, A.; Pospiech, E.; Koller, J.; Kovatsi, L.; Wollstein, A.; Branicki, W.; et al. Global skin colour prediction from DNA. Hum. Genet. 2017, 136, 847–863. [Google Scholar] [CrossRef]
- Hernando, B.; Ibanez, M.V.; Deserio-Cuesta, J.A.; Soria-Navarro, R.; Vilar-Sastre, I.; Martinez-Cadenas, C. Genetic determinants of freckle occurrence in the Spanish population: Towards ephelides prediction from human DNA samples. Forensic Sci. Int. Genet. 2018, 33, 38–47. [Google Scholar] [CrossRef]
- Hysi, P.G.; Valdes, A.M.; Liu, F.; Furlotte, N.A.; Evans, D.M.; Bataille, V.; Visconti, A.; Hemani, G.; McMahon, G.; Ring, S.M.; et al. Genome-wide association meta-analysis of individuals of European ancestry identifies new loci explaining a substantial fraction of hair color variation and heritability. Nat. Genet. 2018, 50, 652–656. [Google Scholar] [CrossRef]
- Pospiech, E.; Chen, Y.; Kukla-Bartoszek, M.; Breslin, K.; Aliferi, A.; Andersen, J.D.; Ballard, D.; Chaitanya, L.; Freire-Aradas, A.; van der Gaag, K.J.; et al. Towards broadening Forensic DNA Phenotyping beyond pigmentation: Improving the prediction of head hair shape from DNA. Forensic Sci. Int. Genet. 2018, 37, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Sun, Y.; Zhao, W.; Gao, X.; Ma, M.; Liu, F.; Li, C. Predicting adult height from DNA variants in a European-Asian admixed population. Int. J. Legal Med. 2019, 133, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Kukla-Bartoszek, M.; Pospiech, E.; Wozniak, A.; Boron, M.; Karlowska-Pik, J.; Teisseyre, P.; Zubanska, M.; Bronikowska, A.; Grzybowski, T.; Ploski, R.; et al. DNA-based predictive models for the presence of freckles. Forensic Sci. Int. Genet. 2019, 42, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhong, K.; Jing, X.; Uitterlinden, A.G.; Hendriks, A.E.J.; Drop, S.L.S.; Kayser, M. Update on the predictability of tall stature from DNA markers in Europeans. Forensic Sci. Int. Genet. 2019, 42, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Liu, F.; Ballantyne, K.N.; van Oven, M.; Lao, O.; Kayser, M. IrisPlex: A sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci. Int. Genet. 2011, 5, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.; Liu, F.; Wollstein, A.; Kovatsi, L.; Ralf, A.; Kosiniak-Kamysz, A.; Branicki, W.; Kayser, M. The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA. Forensic Sci. Int. Genet. 2013, 7, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, L.; Breslin, K.; Zuniga, S.; Wirken, L.; Pospiech, E.; Kukla-Bartoszek, M.; Sijen, T.; Knijff, P.; Liu, F.; Branicki, W.; et al. The HIrisPlex-S system for eye, hair and skin colour prediction from DNA: Introduction and forensic developmental validation. Forensic Sci. Int. Genet. 2018, 35, 123–135. [Google Scholar] [CrossRef]
- Phillips, C.; Salas, A.; Sanchez, J.J.; Fondevila, M.; Gomez-Tato, A.; Alvarez-Dios, J.; Calaza, M.; de Cal, M.C.; Ballard, D.; Lareu, M.V.; et al. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs. Forensic Sci. Int. Genet. 2007, 1, 273–280. [Google Scholar] [CrossRef]
- Kersbergen, P.; van Duijn, K.; Kloosterman, A.D.; den Dunnen, J.T.; Kayser, M.; de Knijff, P. Developing a set of ancestry-sensitive DNA markers reflecting continental origins of humans. BMC Genet. 2009, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Freire Aradas, A.; Kriegel, A.K.; Fondevila, M.; Bulbul, O.; Santos, C.; Serrulla Rech, F.; Perez Carceles, M.D.; Carracedo, A.; Schneider, P.M.; et al. Eurasiaplex: A forensic SNP assay for differentiating European and South Asian ancestries. Forensic Sci. Int. Genet. 2013, 7, 359–366. [Google Scholar] [CrossRef]
- Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Furtado, M.R.; Fang, R.; Madbouly, A.; Maiers, M.; Middha, M.; Friedlaender, F.R.; Kidd, J.R. Progress toward an efficient panel of SNPs for ancestry inference. Forensic Sci. Int. Genet. 2014, 10, 23–32. [Google Scholar] [CrossRef]
- Pereira, V.; Freire-Aradas, A.; Ballard, D.; Borsting, C.; Diez, V.; Pruszkowska-Przybylska, P.; Ribeiro, J.; Achakzai, N.M.; Aliferi, A.; Bulbul, O.; et al. Development and validation of the EUROFORGEN NAME (North African and Middle Eastern) ancestry panel. Forensic Sci. Int. Genet. 2019, 42, 260–267. [Google Scholar] [CrossRef]
- Eduardoff, M.; Gross, T.E.; Santos, C.; de la Puente, M.; Ballard, D.; Strobl, C.; Borsting, C.; Morling, N.; Fusco, L.; Hussing, C.; et al. Inter-laboratory evaluation of the EUROFORGEN Global ancestry-informative SNP panel by massively parallel sequencing using the Ion PGM. Forensic Sci. Int. Genet. 2016, 23, 178–189. [Google Scholar] [CrossRef]
- Mehta, B.; Daniel, R.; Phillips, C.; Doyle, S.; Elvidge, G.; McNevin, D. Massively parallel sequencing of customised forensically informative SNP panels on the MiSeq. Electrophoresis 2016, 37, 2832–2840. [Google Scholar] [CrossRef] [PubMed]
- Breslin, K.; Wills, B.; Ralf, A.; Ventayol Garcia, M.; Kukla-Bartoszek, M.; Pospiech, E.; Freire-Aradas, A.; Xavier, C.; Ingold, S.; de La Puente, M.; et al. HIrisPlex-S system for eye, hair, and skin color prediction from DNA: Massively parallel sequencing solutions for two common forensically used platforms. Forensic Sci. Int. Genet. 2019, 43, 102152. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; de la Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.; Revoir, A.; Pośpiech, E.; Kartasińska, E.; et al. Development and validation of the VISAGE AmpliSeq Basic Tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet 2020, in press. [Google Scholar] [CrossRef]
- Niederstätter, H.; Köchl, S.; Grubwieser, P.; Pavlic, M.; Steinlechner, M.; Parson, W. A modular real-time PCR concept for determining the quantity and quality of human nuclear and mitochondrial DNA. Forensic Sci. Int. Genet. 2007, 1, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.P. 1000 Genomes Project New York Genome Center High Coverage Dataset. Available online: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20190425_NYGC_GATK/ (accessed on 1 May 2020).
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Promega. Prototype PowerSeq SNP System (March 2019); Promega Corporation: Madison, WI, USA, 2019. [Google Scholar]
- Promega. ProNex Size-Selective Purification System. Technical Manual. TM508-2/18; Promega Corporation: Madison, WI, USA, 2018. [Google Scholar]
- Biosystems, K. KAPA Hyper Prep Kit. Technical Data Sheet. KR0961-v6.1; Kapa Biosystems: Cape Town, South Africa, 2017. [Google Scholar]
- Promega. PowerSeq Quant MS System. Technical Manual. TM511-3/18; Promega Corporation: Madison, WI, USA, 2018. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Viena, Austria, 2019. [Google Scholar]
- RStudio. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2018. [Google Scholar]
- Jager, A.C.; Alvarez, M.L.; Davis, C.P.; Guzman, E.; Han, Y.; Way, L.; Walichiewicz, P.; Silva, D.; Pham, N.; Caves, G.; et al. Developmental validation of the MiSeq FGx Forensic Genomics System for Targeted Next Generation Sequencing in Forensic DNA Casework and Database Laboratories. Forensic Sci. Int. Genet. 2017, 28, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Avent, I.; Kinnane, A.G.; Jones, N.; Petermann, I.; Daniel, R.; Gahan, M.E.; McNevin, D. The QIAGEN 140-locus single-nucleotide polymorphism (SNP) panel for forensic identification using massively parallel sequencing (MPS): An evaluation and a direct-to-PCR trial. Int. J. Legal Med. 2019, 133, 677–688. [Google Scholar] [CrossRef] [PubMed]
- de la Puente, M.; Phillips, C.; Xavier, C.; Amigo, J.; Carracedo, A.; Parson, W.; Lareu, M.V. Building a custom large-scale panel of novel microhaplotypes for forensic identification using MiSeq and Ion S5 massively parallel sequencing systems. Forensic Sci. Int. Genet. 2020, 45, 102213. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; Eisenberg, A.J.; van Daal, A. Validity of low copy number typing and applications to forensic science. Croat. Med. J. 2009, 50, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Haned, H.; Bleka, O.; Hansson, O.; Dorum, G.; Egeland, T. Genotyping and interpretation of STR-DNA: Low-template, mixtures and database matches-Twenty years of research and development. Forensic Sci. Int. Genet. 2015, 18, 100–117. [Google Scholar] [CrossRef]
- Buchard, A.; Kampmann, M.L.; Poulsen, L.; Borsting, C.; Morling, N. ISO 17025 validation of a next-generation sequencing assay for relationship testing. Electrophoresis 2016, 37, 2822–2831. [Google Scholar] [CrossRef]
- Eduardoff, M.; Santos, C.; de la Puente, M.; Gross, T.E.; Fondevila, M.; Strobl, C.; Sobrino, B.; Ballard, D.; Schneider, P.M.; Carracedo, A.; et al. Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the Ion PGM. Forensic Sci. Int. Genet. 2015, 17, 110–121. [Google Scholar] [CrossRef]
- Grandell, I.; Samara, R.; Tillmar, A.O. A SNP panel for identity and kinship testing using massive parallel sequencing. Int. J. Legal Med. 2016, 130, 905–914. [Google Scholar] [CrossRef]
- Guo, F.; Yu, J.; Zhang, L.; Li, J. Massively parallel sequencing of forensic STRs and SNPs using the Illumina® ForenSeq DNA Signature Prep Kit on the MiSeq FGx Forensic Genomics System. Forensic Sci. Int. Genet. 2017, 31, 135–148. [Google Scholar] [CrossRef]
- Fattorini, P.; Previdere, C.; Carboni, I.; Marrubini, G.; Sorcaburu-Cigliero, S.; Grignani, P.; Bertoglio, B.; Vatta, P.; Ricci, U. Performance of the ForenSeq(TM) DNA Signature Prep kit on highly degraded samples. Electrophoresis 2017, 38, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.M.; Campbell, N.G.; Cai, G.; Yu, F.; Makarov, V.; Yoon, S.; Daly, M.J.; Gibbs, R.A.; Schellenberg, G.D.; Devlin, B.; et al. Whole exome sequencing reveals minimal differences between cell line and whole blood derived DNA. Genomics 2013, 102, 270–277. [Google Scholar] [CrossRef] [PubMed]
- International Cell Line Authentication Committee (ICLAC). Guide to Human Cell Line Authentication. 2014. Available online: http://iclac.org/wp-content/uploads/Authentication-SOP_09-Jan-2014.pdf (accessed on 11 May 2020).



{kind=link}
{kind=link}
| Sample | 42-S3 | 44-S3 | 45-S2 | 49-S4 | 53-S1 |
|---|---|---|---|---|---|
| Total reads | 329,220 | 406,615 | 492,074 | 361,511 | 260,588 |
| Mean per SNP | 2151 | 2657 | 3216 | 2362 | 1703 |
| SD per SNP | 1351 | 2384 | 2104 | 1491 | 1202 |
| SNPs with < 200 reads | 2 | 11 | 2 | 3 | 9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palencia-Madrid, L.; Xavier, C.; de la Puente, M.; Hohoff, C.; Phillips, C.; Kayser, M.; Parson, W., on behalf of the VISAGE Consortium. Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System. Genes 2020, 11, 708. https://doi.org/10.3390/genes11060708
Palencia-Madrid L, Xavier C, de la Puente M, Hohoff C, Phillips C, Kayser M, Parson W on behalf of the VISAGE Consortium. Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System. Genes. 2020; 11(6):708. https://doi.org/10.3390/genes11060708
Chicago/Turabian StylePalencia-Madrid, Leire, Catarina Xavier, María de la Puente, Carsten Hohoff, Christopher Phillips, Manfred Kayser, and Walther Parson on behalf of the VISAGE Consortium. 2020. "Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System" Genes 11, no. 6: 708. https://doi.org/10.3390/genes11060708
APA StylePalencia-Madrid, L., Xavier, C., de la Puente, M., Hohoff, C., Phillips, C., Kayser, M., & Parson, W., on behalf of the VISAGE Consortium. (2020). Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System. Genes, 11(6), 708. https://doi.org/10.3390/genes11060708