Preimplantation Genetic Testing for Chromosomal Abnormalities: Aneuploidy, Mosaicism, and Structural Rearrangements


{kind=link}
Abstract
1. Introduction
2. Human Reproduction and Chromosomal Abnormalities
3. Overview of Chromosomal Abnormalities in Embryos: Types, Mechanisms, Incidence, and Medical Implications
3.1. Aneuploidy
3.2. Chromosomal Mosaicism
3.3. Segmental Abnormalities
3.4. Structural Rearrangements
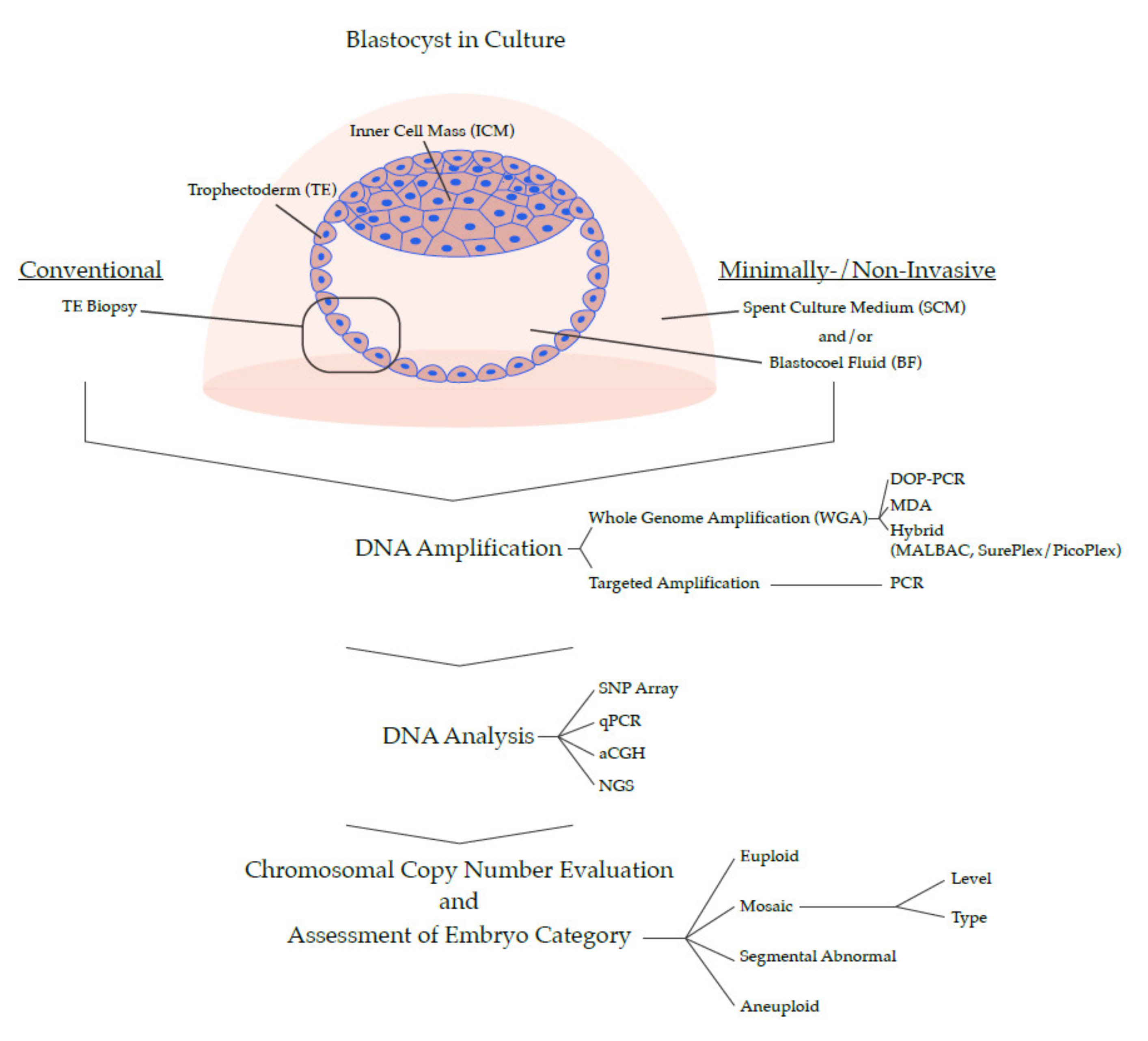
4. Development of PGT for Chromosomal Abnormalities
4.1. Early Methods of PGT-A
4.2. Development of PGT-A for All Chromosomes
4.3. Contemporary PGT-A
5. Considerations on the Clinical Use of PGT for Chromosomal Abnormalities
5.1. How Reliable Is PGT-A?
5.2. Discussion on the Clinical Merits of PGT-A
5.3. A Note on the STAR Study
5.4. Which Patients and Embryos Should Be Offered PGT-A?
6. Refinement of PGT-A Categories
6.1. Management of Mosaic Embryos in the Clinic
6.2. Why Segmental Abnormality Should Be Managed Differently
6.3. Refinement of the PGT-A Category System: Is it Necessary?
7. Current Developments and Future Directions of PGT for Chromosomal Abnormalities
7.1. Mitochondrial DNA Quantitation During PGT-A: Where Are We Now
7.2. Variations and Add-Ons to Conventional PGT-A
7.3. The Prospect of Non-Invasive PGT-A
8. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Bromer, J.G.; Ata, B.; Seli, M.; Lockwood, C.J.; Seli, E. Preterm deliveries that result from multiple pregnancies associated with assisted reproductive technologies in the USA: A cost analysis. Curr. Opin. Obs. Gynecol. 2011, 23, 168–173. [Google Scholar] [CrossRef]
- Murray, S.R.; Norman, J.E. Multiple pregnancies following assisted reproductive technologies—A happy consequence or double trouble? Semin. Fetal Neonatal Med. 2014, 19, 222–227. [Google Scholar] [CrossRef]
- Hill, G.A.; Freeman, M.; Bastias, M.C.; Rogers, B.J.; Herbert, C.M., 3rd; Osteen, K.G.; Wentz, A.C. The influence of oocyte maturity and embryo quality on pregnancy rate in a program for in vitro fertilization-embryo transfer. Fertil. Steril. 1989, 52, 801–806. [Google Scholar]
- Gardner, D.K.; Schoolcraft, W.B. In vitro culture of human blastocysts. In Towards Reproductive Certainty: Fertility and Genetics Beyond; Jansen, R., Mortimer, D., Eds.; Parthenon Publishing: Nashville, TN, USA, 1999; pp. 378–388. [Google Scholar]
- Capalbo, A.; Rienzi, L.; Cimadomo, D.; Maggiulli, R.; Elliott, T.; Wright, G.; Nagy, Z.P.; Ubaldi, F.M. Correlation between standard blastocyst morphology, euploidy and implantation: An observational study in two centers involving 956 screened blastocysts. Hum. Reprod. 2014, 29, 1173–1181. [Google Scholar] [CrossRef]
- Handyside, A.H.; Kontogianni, E.H.; Hardy, K.; Winston, R.M. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 1990, 344, 768–770. [Google Scholar]
- International Federation of Fertility Societies’ Surveillance (IFFS) 2019: Global Trends in Reproductive Policy and Practice, 8th Edition. Glob. Reprod. Health 2019, 4, e29. [CrossRef]
- Gruhn, J.R.; Zielinska, A.P.; Shukla, V.; Blanshard, R.; Capalbo, A.; Cimadomo, D.; Nikiforov, D.; Chan, A.C.; Newnham, L.J.; Vogel, I.; et al. Chromosome errors in human eggs shape natural fertility over reproductive life span. Science 2019, 365, 1466–1469. [Google Scholar]
- Hawkes, K.; Smith, K.R. Do women stop early? Similarities in fertility decline in humans and chimpanzees. Ann. N. Y. Acad. Sci. 2010, 1204, 43–53. [Google Scholar] [CrossRef]
- Franasiak, J.M.; Forman, E.J.; Hong, K.H.; Werner, M.D.; Upham, K.M.; Treff, N.R.; Scott, R.T. Aneuploidy across individual chromosomes at the embryonic level in trophectoderm biopsies: Changes with patient age and chromosome structure. J. Assist. Reprod. Genet. 2014, 31, 1501–1509. [Google Scholar]
- Rubio, C.; Rodrigo, L.; Garcia-Pascual, C.; Peinado, V.; Campos-Galindo, I.; Garcia-Herrero, S.; Simon, C. Clinical application of embryo aneuploidy testing by next-generation sequencing. Biol. Reprod. 2019, 101, 1083–1090. [Google Scholar] [CrossRef]
- Taylor, T.H.; Gitlin, S.A.; Patrick, J.L.; Crain, J.L.; Wilson, J.M.; Griffin, D.K. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 2014, 20, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.J.; Chandley, A.C. Aneuploidy: The Origins and Causes of Aneuploidy in Experimental Organisms. In Aneuploidy; Bond, D.J., Chandley, A.C., Eds.; Oxford University Press: Oxford, UK, 1983; pp. 27–54. [Google Scholar]
- Irani, M.; Zaninovic, N.; Rosenwaks, Z.; Xu, K. Does maternal age at retrieval influence the implantation potential of euploid blastocysts? Am. J. Obs. Gynecol. 2019, 220, 379 e1–379 e7. [Google Scholar] [CrossRef]
- Harton, G.L.; Munné, S.; Surrey, M.; Grifo, J.; Kaplan, B.; McCulloh, D.H.; Griffin, D.K.; Wells, D.; Group, P.G.D.P. Diminished effect of maternal age on implantation after preimplantation genetic diagnosis with array comparative genomic hybridization. Fertil. Steril. 2013, 100, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, D.; Hornak, M.; Horak, J.; Navratil, R.; Tauwinklova, G.; Rubes, J.; Vesela, K. Incidence and origin of meiotic whole and segmental chromosomal aneuploidies detected by karyomapping. Reprod. Biomed. Online 2019, 38, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, M.; Ravichandran, K.; Gunes, Z.; Prates, R.; Goodall, N.N.; Roman, B.; Ribustello, L.; Shanmugam, A.; Colls, P.; Munne, S.; et al. Aneuploidy and recombination in the human preimplantation embryo. Copy number variation analysis and genome-wide polymorphism genotyping. Reprod. Biomed. Online 2020, 40, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Bono, S.; Spizzichino, L.; Biricik, A.; Baldi, M.; Colamaria, S.; Ubaldi, F.M.; Rienzi, L.; Fiorentino, F. Sequential comprehensive chromosome analysis on polar bodies, blastomeres and trophoblast: Insights into female meiotic errors and chromosomal segregation in the preimplantation window of embryo development. Hum. Reprod. 2013, 28, 509–518. [Google Scholar]
- Ottolini, C.S.; Newnham, L.; Capalbo, A.; Natesan, S.A.; Joshi, H.A.; Cimadomo, D.; Griffin, D.K.; Sage, K.; Summers, M.C.; Thornhill, A.R.; et al. Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat. Genet. 2015, 47, 727–735. [Google Scholar] [CrossRef]
- Martin, R.H.; Rademaker, A. The frequency of aneuploidy among individual chromosomes in 6,821 human sperm chromosome complements. Cytogenet. Cell Genet. 1990, 53, 103–107. [Google Scholar] [CrossRef]
- Templado, C.; Vidal, F.; Estop, A. Aneuploidy in human spermatozoa. Cytogenet. Genome Res. 2011, 133, 91–99. [Google Scholar]
- Grati, F.R.; Gallazzi, G.; Branca, L.; Maggi, F.; Simoni, G.; Yaron, Y. An evidence-based scoring system for prioritizing mosaic aneuploid embryos following preimplantation genetic screening. Reprod. Biomed. Online 2018, 36, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.; Abruzzo, M.; Adkins, K.; Griffin, D.; Merrill, M.; Millie, E.; Saker, D.; Shen, J.; Zaragoza, M. Human aneuploidy: Incidence, origin, and etiology. Env. Mol. Mutagen. 1996, 28, 167–175. [Google Scholar] [CrossRef]
- Munné, S.; Alikani, M.; Ribustello, L.; Colls, P.; Martinez-Ortiz, P.A.; McCulloh, D.H.; Referring Physician, G. Euploidy rates in donor egg cycles significantly differ between fertility centers. Hum. Reprod. 2017, 32, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Demko, Z.P.; Simon, A.L.; McCoy, R.C.; Petrov, D.A.; Rabinowitz, M. Effects of maternal age on euploidy rates in a large cohort of embryos analyzed with 24-chromosome single-nucleotide polymorphism-based preimplantation genetic screening. Fertil. Steril. 2016, 105, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Franasiak, J.M.; Forman, E.J.; Hong, K.H.; Werner, M.D.; Upham, K.M.; Treff, N.R.; Scott, R.T., Jr. The nature of aneuploidy with increasing age of the female partner: A review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil. Steril. 2014, 101, 656–663. [Google Scholar] [CrossRef]
- Irani, M.; Canon, C.; Robles, A.; Maddy, B.; Gunnala, V.; Qin, X.; Zhang, C.; Xu, K.; Rosenwaks, Z. No effect of ovarian stimulation and oocyte yield on euploidy and live birth rates: An analysis of 12 298 trophectoderm biopsies. Hum. Reprod. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.; Schultz, R.M.; Lampson, M.A. Meiotic origins of maternal age-related aneuploidy. Biol. Reprod. 2012, 86, 1–7. [Google Scholar] [CrossRef]
- Capalbo, A.; Hoffmann, E.R.; Cimadomo, D.; Ubaldi, F.M.; Rienzi, L. Human female meiosis revised: New insights into the mechanisms of chromosome segregation and aneuploidies from advanced genomics and time-lapse imaging. Hum. Reprod. Update 2017, 23, 706–722. [Google Scholar] [CrossRef]
- Carrasquillo, R.J.; Kohn, T.P.; Cinnioglu, C.; Rubio, C.; Simon, C.; Ramasamy, R.; Al-Asmar, N. Advanced paternal age does not affect embryo aneuploidy following blastocyst biopsy in egg donor cycles. J. Assist. Reprod. Genet. 2019, 36, 2039–2045. [Google Scholar] [CrossRef]
- Baart, E.B.; Martini, E.; Eijkemans, M.J.; Van Opstal, D.; Beckers, N.G.; Verhoeff, A.; Macklon, N.S.; Fauser, B.C. Milder ovarian stimulation for in-vitro fertilization reduces aneuploidy in the human preimplantation embryo: A randomized controlled trial. Hum. Reprod. 2007, 22, 980–988. [Google Scholar] [CrossRef]
- Rubio, C.; Mercader, A.; Alama, P.; Lizan, C.; Rodrigo, L.; Labarta, E.; Melo, M.; Pellicer, A.; Remohi, J. Prospective cohort study in high responder oocyte donors using two hormonal stimulation protocols: Impact on embryo aneuploidy and development. Hum. Reprod. 2010, 25, 2290–2297. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Amon, A. The aneuploidy paradox: Costs and benefits of an incorrect karyotype. Trends Genet. 2011, 27, 446–453. [Google Scholar] [CrossRef]
- Fragouli, E.; Alfarawati, S.; Spath, K.; Babariya, D.; Tarozzi, N.; Borini, A.; Wells, D. Analysis of implantation and ongoing pregnancy rates following the transfer of mosaic diploid-aneuploid blastocysts. Hum. Genet. 2017, 136, 805–819. [Google Scholar] [CrossRef]
- Greco, E.; Minasi, M.G.; Fiorentino, F. Healthy Babies after Intrauterine Transfer of Mosaic Aneuploid Blastocysts. N. Engl. J. Med. 2015, 373, 2089–2090. [Google Scholar] [CrossRef]
- Munné, S.; Blazek, J.; Large, M.; Martinez-Ortiz, P.A.; Nisson, H.; Liu, E.; Tarozzi, N.; Borini, A.; Becker, A.; Zhang, J.; et al. Detailed investigation into the cytogenetic constitution and pregnancy outcome of replacing mosaic blastocysts detected with the use of high-resolution next-generation sequencing. Fertil. Steril. 2017, 108, 62–71. [Google Scholar] [CrossRef]
- Munné, S.; Spinella, F.; Grifo, J.; Zhang, J.; Beltran, M.P.; Fragouli, E.; Fiorentino, F. Clinical outcomes after the transfer of blastocysts characterized as mosaic by high resolution Next Generation Sequencing-further insights. Eur. J. Med. Genet. 2020, 63, 103741. [Google Scholar] [CrossRef]
- Spinella, F.; Fiorentino, F.; Biricik, A.; Bono, S.; Ruberti, A.; Cotroneo, E.; Baldi, M.; Cursio, E.; Minasi, M.G.; Greco, E. Extent of chromosomal mosaicism influences the clinical outcome of in vitro fertilization treatments. Fertil. Steril. 2018, 109, 77–83. [Google Scholar] [CrossRef]
- Victor, A.R.; Tyndall, J.C.; Brake, A.J.; Lepkowsky, L.T.; Murphy, A.E.; Griffin, D.K.; McCoy, R.C.; Barnes, F.L.; Zouves, C.G.; Viotti, M. One hundred mosaic embryos transferred prospectively in a single clinic: Exploring when and why they result in healthy pregnancies. Fertil. Steril. 2019, 111, 280–293. [Google Scholar] [CrossRef]
- Vazquez-Diez, C.; FitzHarris, G. Causes and consequences of chromosome segregation error in preimplantation embryos. Reproduction 2018, 155, R63–R76. [Google Scholar] [CrossRef]
- Victor, A.; Ogur, C.; Thornhill, A.; Griffin, D. Preimplantation Genetic Testing for Aneuploidies: Where We Are and Where We’re Going. In Preimplantation Genetic Testing: Recent Advances in Reproductive Medicine, 2020 ed.; Griffin, D., Harton, G., Eds.; Taylor & Francis Group: Abingdon, UK, 2020. [Google Scholar]
- Coonen, E.; Derhaag, J.G.; Dumoulin, J.C.; van Wissen, L.C.; Bras, M.; Janssen, M.; Evers, J.L.; Geraedts, J.P. Anaphase lagging mainly explains chromosomal mosaicism in human preimplantation embryos. Hum. Reprod. 2004, 19, 316–324. [Google Scholar] [CrossRef]
- Ioannou, D.; Fonseka, K.G.; Meershoek, E.J.; Thornhill, A.R.; Abogrein, A.; Ellis, M.; Griffin, D.K. Twenty-four chromosome FISH in human IVF embryos reveals patterns of post-zygotic chromosome segregation and nuclear organisation. Chromosome Res. 2012, 20, 447–460. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.C. Mosaicism in Preimplantation Human Embryos: When Chromosomal Abnormalities Are the Norm. Trends Genet. 2017, 33, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hao, Y.; Elshewy, N.; Zhu, X.; Zhang, Z.; Zhou, P. The mechanisms and clinical application of mosaicism in preimplantation embryos. J. Assist. Reprod. Genet. 2020, 37, 497–508. [Google Scholar] [PubMed]
- Delhanty, J.D.; Griffin, D.K.; Handyside, A.H.; Harper, J.; Atkinson, G.H.; Pieters, M.H.; Winston, R.M. Detection of aneuploidy and chromosomal mosaicism in human embryos during preimplantation sex determination by fluorescent in situ hybridisation, (FISH). Hum. Mol. Genet. 1993, 2, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Evsikov, S.; Verlinsky, Y. Mosaicism in the inner cell mass of human blastocysts. Hum. Reprod. 1998, 13, 3151–3155. [Google Scholar] [CrossRef]
- Popovic, M.; Dhaenens, L.; Boel, A.; Menten, B.; Heindryckx, B. Chromosomal mosaicism in human blastocysts: The ultimate diagnostic dilemma. Hum. Reprod. Update 2020, 26, 313–334. [Google Scholar] [CrossRef]
- Munné, S.; Kaplan, B.; Frattarelli, J.L.; Child, T.; Nakhuda, G.; Shamma, F.N.; Silverberg, K.; Kalista, T.; Handyside, A.H.; Katz-Jaffe, M.; et al. Preimplantation genetic testing for aneuploidy versus morphology as selection criteria for single frozen-thawed embryo transfer in good-prognosis patients: A multicenter randomized clinical trial. Fertil. Steril. 2019, 112, 1071–1079. [Google Scholar]
- Munné, S.; Wells, D. Detection of mosaicism at blastocyst stage with the use of high-resolution next-generation sequencing. Fertil. Steril. 2017, 107, 1085–1091. [Google Scholar] [CrossRef]
- Tsuiko, O.; Catteeuw, M.; Zamani Esteki, M.; Destouni, A.; Bogado Pascottini, O.; Besenfelder, U.; Havlicek, V.; Smits, K.; Kurg, A.; Salumets, A.; et al. Genome stability of bovine in vivo-conceived cleavage-stage embryos is higher compared to in vitro-produced embryos. Hum. Reprod. 2017, 32, 2348–2357. [Google Scholar] [CrossRef]
- Zamani Esteki, M.; Viltrop, T.; Tsuiko, O.; Tiirats, A.; Koel, M.; Noukas, M.; Zilina, O.; Teearu, K.; Marjonen, H.; Kahila, H.; et al. In vitro fertilization does not increase the incidence of de novo copy number alterations in fetal and placental lineages. Nat. Med. 2019, 25, 1699–1705. [Google Scholar]
- McCoy, R.C.; Demko, Z.P.; Ryan, A.; Banjevic, M.; Hill, M.; Sigurjonsson, S.; Rabinowitz, M.; Petrov, D.A. Evidence of Selection against Complex Mitotic-Origin Aneuploidy during Preimplantation Development. PLoS Genet. 2015, 11, e1005601. [Google Scholar] [CrossRef]
- Vera-Rodriguez, M.; Rubio, C. Assessing the true incidence of mosaicism in preimplantation embryos. Fertil. Steril. 2017, 107, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Bolton, H.; Graham, S.J.; Van der Aa, N.; Kumar, P.; Theunis, K.; Fernandez Gallardo, E.; Voet, T.; Zernicka-Goetz, M. Mouse model of chromosome mosaicism reveals lineage-specific depletion of aneuploid cells and normal developmental potential. Nat. Commun. 2016, 7, 11165. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, D.; Ikeda, Z.; Yao, T.; Tokoro, M.; Fukunaga, N.; Asada, Y.; Yamagata, K. Chromosome segregation error during early cleavage in mouse pre-implantation embryo does not necessarily cause developmental failure after blastocyst stage. Sci. Rep. 2020, 10, 854. [Google Scholar] [PubMed]
- Popovic, M.; Dhaenens, L.; Taelman, J.; Dheedene, A.; Bialecka, M.; De Sutter, P.; Chuva de Sousa Lopes, S.M.; Menten, B.; Heindryckx, B. Extended in vitro culture of human embryos demonstrates the complex nature of diagnosing chromosomal mosaicism from a single trophectoderm biopsy. Hum. Reprod. 2019, 34, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Kuliev, A.; Verlinsky, Y. Meiotic and mitotic nondisjunction: Lessons from preimplantation genetic diagnosis. Hum. Reprod. Update 2004, 10, 401–407. [Google Scholar] [CrossRef][Green Version]
- Gueye, N.A.; Devkota, B.; Taylor, D.; Pfundt, R.; Scott, R.T., Jr.; Treff, N.R. Uniparental disomy in the human blastocyst is exceedingly rare. Fertil. Steril. 2014, 101, 232–236. [Google Scholar] [CrossRef]
- Fryburg, J.S.; Dimaio, M.S.; Yang-Feng, T.L.; Mahoney, M.J. Follow-up of pregnancies complicated by placental mosaicism diagnosed by chorionic villus sampling. Prenat. Diagn. 1993, 13, 481–494. [Google Scholar]
- Kalousek, D.K.; Dill, F.J. Chromosomal mosaicism confined to the placenta in human conceptions. Science 1983, 221, 665–667. [Google Scholar] [CrossRef]
- Leschot, N.J.; Schuring-Blom, G.H.; Van Prooijen-Knegt, A.C.; Verjaal, M.; Hansson, K.; Wolf, H.; Kanhai, H.H.; Van Vugt, J.M.; Christiaens, G.C. The outcome of pregnancies with confined placental chromosome mosaicism in cytotrophoblast cells. Prenat. Diagn. 1996, 16, 705–712. [Google Scholar]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016, 17, 241. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Kutsev, S.I.; Pellestor, F.; Beresheva, A.K.; Demidova, I.A.; Kravets, V.S.; et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE 2007, 2, e558. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Biricik, A.; Bono, S.; Spizzichino, L.; Cotroneo, E.; Cottone, G.; Kokocinski, F.; Michel, C.E. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil. Steril. 2014, 101, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Tan, K.; Vajta, G.; Jiang, H.; Tan, Y.; Zhang, C.; Chen, F.; Chen, S.; Zhang, C.; Pan, X.; et al. Massively parallel sequencing for chromosomal abnormality testing in trophectoderm cells of human blastocysts. Biol. Reprod. 2013, 88, 69. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jin, H.; Liu, L.; Liu, J.; Wang, W.H. Application of next-generation sequencing for 24-chromosome aneuploidy screening of human preimplantation embryos. Mol. Cytogenet. 2015, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Babariya, D.; Fragouli, E.; Alfarawati, S.; Spath, K.; Wells, D. The incidence and origin of segmental aneuploidy in human oocytes and preimplantation embryos. Hum. Reprod. 2017, 32, 2549–2560. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Richardson, C.; Horikoshi, N.; Pandita, T.K. The role of the DNA double-strand break response network in meiosis. DNA Repair 2004, 3, 1149–1164. [Google Scholar] [CrossRef]
- Aguilera, A.; Garcia-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef]
- Escriba, M.J.; Vendrell, X.; Peinado, V. Segmental aneuploidy in human blastocysts: A qualitative and quantitative overview. Reprod. Biol. Endocrinol. 2019, 17, 76. [Google Scholar] [CrossRef] [PubMed]
- Girardi, L.; Serdarogullari, M.; Patassini, C.; Poli, M.; Fabiani, M.; Caroselli, S.; Coban, O.; Findikli, N.; Boynukalin, F.K.; Bahceci, M.; et al. Incidence, Origin, and Predictive Model for the Detection and Clinical Management of Segmental Aneuploidies in Human Embryos. Am. J. Hum. Genet. 2020, 106, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Vera-Rodriguez, M.; Michel, C.E.; Mercader, A.; Bladon, A.J.; Rodrigo, L.; Kokocinski, F.; Mateu, E.; Al-Asmar, N.; Blesa, D.; Simon, C.; et al. Distribution patterns of segmental aneuploidies in human blastocysts identified by next-generation sequencing. Fertil. Steril. 2016, 105, 1047–1055. [Google Scholar] [PubMed]
- Martinez, M.C.; Mendez, C.; Ferro, J.; Nicolas, M.; Serra, V.; Landeras, J. Cytogenetic analysis of early nonviable pregnancies after assisted reproduction treatment. Fertil. Steril. 2010, 93, 289–292. [Google Scholar]
- Wellesley, D.; Dolk, H.; Boyd, P.A.; Greenlees, R.; Haeusler, M.; Nelen, V.; Garne, E.; Khoshnood, B.; Doray, B.; Rissmann, A.; et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur. J. Hum. Genet. 2012, 20, 521–526. [Google Scholar]
- Shaffer, L.G.; Lupski, J.R. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu. Rev. Genet. 2000, 34, 297–329. [Google Scholar] [CrossRef]
- Chow, J.F.C.; Cheng, H.H.Y.; Lau, E.Y.L.; Yeung, W.S.B.; Ng, E.H.Y. Distinguishing between carrier and noncarrier embryos with the use of long-read sequencing in preimplantation genetic testing for reciprocal translocations. Genomics 2020, 112, 494–500. [Google Scholar] [CrossRef]
- Treff, N.R.; Thompson, K.; Rafizadeh, M.; Chow, M.; Morrison, L.; Tao, X.; Garnsey, H.; Reda, C.V.; Metzgar, T.L.; Neal, S.; et al. SNP array-based analyses of unbalanced embryos as a reference to distinguish between balanced translocation carrier and normal blastocysts. J. Assist. Reprod. Genet. 2016, 33, 1115–1119. [Google Scholar]
- Wang, L.; Shen, J.; Cram, D.S.; Ma, M.; Wang, H.; Zhang, W.; Fan, J.; Gao, Z.; Zhang, L.; Li, Z.; et al. Preferential selection and transfer of euploid noncarrier embryos in preimplantation genetic diagnosis cycles for reciprocal translocations. Fertil. Steril. 2017, 108, 620–627. [Google Scholar]
- Xu, J.; Zhang, Z.; Niu, W.; Yang, Q.; Yao, G.; Shi, S.; Jin, H.; Song, W.; Chen, L.; Zhang, X.; et al. Mapping allele with resolved carrier status of Robertsonian and reciprocal translocation in human preimplantation embryos. Proc. Natl. Acad. Sci. USA 2017, 114, E8695–E8702. [Google Scholar] [CrossRef]
- Ferfouri, F.; Bernicot, I.; Schneider, A.; Haquet, E.; Hedon, B.; Anahory, T. Is the resulting phenotype of an embryo with balanced X-autosome translocation, obtained by means of preimplantation genetic diagnosis, linked to the X inactivation pattern? Fertil. Steril. 2016, 105, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.K.; Wilton, L.J.; Handyside, A.H.; Atkinson, G.H.; Winston, R.M.; Delhanty, J.D. Diagnosis of sex in preimplantation embryos by fluorescent in situ hybridisation. BMJ 1993, 306, 1382. [Google Scholar] [PubMed]
- Munné, S.; Lee, A.; Rosenwaks, Z.; Grifo, J.; Cohen, J. Diagnosis of major chromosome aneuploidies in human preimplantation embryos. Hum. Reprod. 1993, 8, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- Schrurs, B.M.; Winston, R.M.; Handyside, A.H. Preimplantation diagnosis of aneuploidy using fluorescent in-situ hybridization: Evaluation using a chromosome 18-specific probe. Hum. Reprod. 1993, 8, 296–301. [Google Scholar] [PubMed]
- Munné, S.; Fragouli, E.; Colls, P.; Katz-Jaffe, M.; Schoolcraft, W.; Wells, D. Improved detection of aneuploid blastocysts using a new 12-chromosome FISH test. Reprod. Biomed. Online 2010, 20, 92–97. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Griffin, D.K.; Ogur, C. Chromosomal analysis in IVF: Just how useful is it? Reproduction 2018, 156, F29–F50. [Google Scholar] [CrossRef]
- Scott, R.T., Jr.; Upham, K.M.; Forman, E.J.; Zhao, T.; Treff, N.R. Cleavage-stage biopsy significantly impairs human embryonic implantation potential while blastocyst biopsy does not: A randomized and paired clinical trial. Fertil. Steril. 2013, 100, 624–630. [Google Scholar] [CrossRef]
- Verlinsky, Y.; Ginsberg, N.; Lifchez, A.; Valle, J.; Moise, J.; Strom, C.M. Analysis of the first polar body: Preconception genetic diagnosis. Hum. Reprod. 1990, 5, 826–829. [Google Scholar]
- Salvaggio, C.N.; Forman, E.J.; Garnsey, H.M.; Treff, N.R.; Scott, R.T., Jr. Polar body based aneuploidy screening is poorly predictive of embryo ploidy and reproductive potential. J. Assist. Reprod. Genet. 2014, 31, 1221–1226. [Google Scholar] [CrossRef][Green Version]
- Levin, I.; Almog, B.; Shwartz, T.; Gold, V.; Ben-Yosef, D.; Shaubi, M.; Amit, A.; Malcov, M. Effects of laser polar-body biopsy on embryo quality. Fertil. Steril. 2012, 97, 1085–1088. [Google Scholar]
- Mastenbroek, S.; Twisk, M.; van der Veen, F.; Repping, S. Preimplantation genetic screening: A systematic review and meta-analysis of RCTs. Hum. Reprod. Update 2011, 17, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Deleye, L.; De Coninck, D.; Christodoulou, C.; Sante, T.; Dheedene, A.; Heindryckx, B.; Van den Abbeel, E.; De Sutter, P.; Menten, B.; Deforce, D.; et al. Whole genome amplification with SurePlex results in better copy number alteration detection using sequencing data compared to the MALBAC method. Sci. Rep. 2015, 5, 11711. [Google Scholar] [PubMed]
- Wells, D.; Delhanty, J.D. Comprehensive chromosomal analysis of human preimplantation embryos using whole genome amplification and single cell comparative genomic hybridization. Mol. Hum. Reprod. 2000, 6, 1055–1062. [Google Scholar]
- Schoolcraft, W.B.; Fragouli, E.; Stevens, J.; Munné, S.; Katz-Jaffe, M.G.; Wells, D. Clinical application of comprehensive chromosomal screening at the blastocyst stage. Fertil. Steril. 2010, 94, 1700–1706. [Google Scholar] [CrossRef]
- Hellani, A.; Abu-Amero, K.; Azouri, J.; El-Akoum, S. Successful pregnancies after application of array-comparative genomic hybridization in PGS-aneuploidy screening. Reprod. Biomed. Online 2008, 17, 841–847. [Google Scholar] [CrossRef]
- Gutierrez-Mateo, C.; Colls, P.; Sanchez-Garcia, J.; Escudero, T.; Prates, R.; Ketterson, K.; Wells, D.; Munne, S. Validation of microarray comparative genomic hybridization for comprehensive chromosome analysis of embryos. Fertil. Steril. 2011, 95, 953–958. [Google Scholar] [CrossRef]
- Treff, N.R.; Su, J.; Tao, X.; Levy, B.; Scott, R.T., Jr. Accurate single cell 24 chromosome aneuploidy screening using whole genome amplification and single nucleotide polymorphism microarrays. Fertil. Steril. 2010, 94, 2017–2021. [Google Scholar]
- Wells, D.; Kaur, K.; Grifo, J.; Glassner, M.; Taylor, J.C.; Fragouli, E.; Munné, S. Clinical utilisation of a rapid low-pass whole genome sequencing technique for the diagnosis of aneuploidy in human embryos prior to implantation. J. Med. Genet. 2014, 51, 553–562. [Google Scholar]
- Treff, N.R.; Tao, X.; Ferry, K.M.; Su, J.; Taylor, D.; Scott, R.T., Jr. Development and validation of an accurate quantitative real-time polymerase chain reaction-based assay for human blastocyst comprehensive chromosomal aneuploidy screening. Fertil. Steril. 2012, 97, 819–824. [Google Scholar]
- Zimmerman, R.S.; Tao, X.; Marin, D.; Werner, M.D.; Hong, K.H.; Lonczak, A.; Landis, J.; Taylor, D.; Zhan, Y.; Scott, R.T., Jr.; et al. Preclinical validation of a targeted next generation sequencing-based comprehensive chromosome screening methodology in human blastocysts. Mol. Hum. Reprod. 2018, 24, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Dokras, A.; Sargent, I.L.; Ross, C.; Gardner, R.L.; Barlow, D.H. Trophectoderm biopsy in human blastocysts. Hum. Reprod. 1990, 5, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Romanelli, V.; Cimadomo, D.; Girardi, L.; Stoppa, M.; Dovere, L.; Dell’Edera, D.; Ubaldi, F.M.; Rienzi, L. Implementing PGD/PGD-A in IVF clinics: Considerations for the best laboratory approach and management. J. Assist. Reprod. Genet. 2016, 33, 1279–1286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McArthur, S.J.; Leigh, D.; Marshall, J.T.; de Boer, K.A.; Jansen, R.P. Pregnancies and live births after trophectoderm biopsy and preimplantation genetic testing of human blastocysts. Fertil. Steril. 2005, 84, 1628–1636. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.H.; Stankewicz, T.; Katz, S.L.; Patrick, J.L.; Johnson, L.; Griffin, D.K. Preliminary assessment of aneuploidy rates between the polar, mid and mural trophectoderm. Zygote 2019, 1–4. [Google Scholar] [CrossRef]
- Romanelli, V.; Poli, M.; Capalbo, A. Preimplantation genetic testing in assisted reproductive technology. Panminerva Med. 2019, 61, 30–41. [Google Scholar] [CrossRef]
- Rubino, P.; Tapia, L.; Ruiz de Assin Alonso, R.; Mazmanian, K.; Guan, L.; Dearden, L.; Thiel, A.; Moon, C.; Kolb, B.; Norian, J.M.; et al. Trophectoderm biopsy protocols can affect clinical outcomes: Time to focus on the blastocyst biopsy technique. Fertil. Steril. 2020, 113, 981–989. [Google Scholar] [CrossRef]
- Cimadomo, D.; Rienzi, L.; Capalbo, A.; Rubio, C.; Innocenti, F.; Garcia-Pascual, C.M.; Ubaldi, F.M.; Handyside, A. The dawn of the future: 30 years from the first biopsy of a human embryo. The detailed history of an ongoing revolution. Hum. Reprod. Update 2020. [Google Scholar] [CrossRef]
- Marek, D.; Langley, M.; Gardner, D.K.; Confer, N.; Doody, K.M.; Doody, K.J. Introduction of blastocyst culture and transfer for all patients in an in vitro fertilization program. Fertil. Steril. 1999, 72, 1035–1040. [Google Scholar] [CrossRef]
- Quinn, P. Enhanced results in mouse and human embryo culture using a modified human tubal fluid medium lacking glucose and phosphate. J. Assist. Reprod. Genet. 1995, 12, 97–105. [Google Scholar] [CrossRef]
- Kuwayama, M. Highly efficient vitrification for cryopreservation of human oocytes and embryos: The Cryotop method. Theriogenology 2007, 67, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Harton, G.L.; De Rycke, M.; Fiorentino, F.; Moutou, C.; SenGupta, S.; Traeger-Synodinos, J.; Harper, J.C.; European Society for Human, R.; Embryology, P.G.D.C. ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum. Reprod. 2011, 26, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Ubaldi, F.M.; Rienzi, L.; Scott, R.; Treff, N. Detecting mosaicism in trophectoderm biopsies: Current challenges and future possibilities. Hum. Reprod. 2017, 32, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Lenzi, M.; Ross, R.; Katz-Jaffe, M.; Schoolcraft, W.B.; Wells, D. Comprehensive molecular cytogenetic analysis of the human blastocyst stage. Hum. Reprod. 2008, 23, 2596–2608. [Google Scholar] [CrossRef] [PubMed]
- Cimadomo, D.; Rienzi, L.; Romanelli, V.; Alviggi, E.; Levi-Setti, P.E.; Albani, E.; Dusi, L.; Papini, L.; Livi, C.; Benini, F.; et al. Inconclusive chromosomal assessment after blastocyst biopsy: Prevalence, causative factors and outcomes after re-biopsy and re-vitrification. A multicenter experience. Hum. Reprod. 2018, 33, 1839–1846. [Google Scholar] [CrossRef]
- Chaubey, A.; Shenoy, S.; Mathur, A.; Ma, Z.; Valencia, C.A.; Reddy Nallamilli, B.R.; Szekeres, E., Jr.; Stansberry, L.; Liu, R.; Hegde, M.R. Low-Pass Genome Sequencing: Validation and Diagnostic Utility from 409 Clinical Cases of Low-Pass Genome Sequencing for the Detection of Copy Number Variants to Replace Constitutional Microarray. J. Mol. Diagn. 2020, 20, 1525–1578. [Google Scholar] [CrossRef]
- Ruttanajit, T.; Chanchamroen, S.; Cram, D.S.; Sawakwongpra, K.; Suksalak, W.; Leng, X.; Fan, J.; Wang, L.; Yao, Y.; Quangkananurug, W. Detection and quantitation of chromosomal mosaicism in human blastocysts using copy number variation sequencing. Prenat. Diagn. 2016, 36, 154–162. [Google Scholar] [CrossRef]
- Friedenthal, J.; Maxwell, S.M.; Munné, S.; Kramer, Y.; McCulloh, D.H.; McCaffrey, C.; Grifo, J.A. Next generation sequencing for preimplantation genetic screening improves pregnancy outcomes compared with array comparative genomic hybridization in single thawed euploid embryo transfer cycles. Fertil. Steril. 2018, 109, 627–632. [Google Scholar] [CrossRef]
- Friedenthal, J.; Maxwell, S.M.; Tiegs, A.W.; Besser, A.G.; McCaffrey, C.; Munné, S.; Noyes, N.; Grifo, J.A. Clinical error rates of next generation sequencing and array comparative genomic hybridization with single thawed euploid embryo transfer. Eur. J. Med. Genet. 2020, 63, 103852. [Google Scholar] [CrossRef]
- Maxwell, S.M.; Colls, P.; Hodes-Wertz, B.; McCulloh, D.H.; McCaffrey, C.; Wells, D.; Munné, S.; Grifo, J.A. Why do euploid embryos miscarry? A case-control study comparing the rate of aneuploidy within presumed euploid embryos that resulted in miscarriage or live birth using next-generation sequencing. Fertil. Steril. 2016, 106, 1414–1419. [Google Scholar] [CrossRef]
- Deleye, L.; Dheedene, A.; De Coninck, D.; Sante, T.; Christodoulou, C.; Heindryckx, B.; Van den Abbeel, E.; De Sutter, P.; Deforce, D.; Menten, B.; et al. Shallow whole genome sequencing is well suited for the detection of chromosomal aberrations in human blastocysts. Fertil. Steril. 2015, 104, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Cram, D.S.; Leigh, D.; Handyside, A.; Rechitsky, L.; Xu, K.; Harton, G.; Grifo, J.; Rubio, C.; Fragouli, E.; Kahraman, S.; et al. PGDIS Position Statement on the Transfer of Mosaic Embryos 2019. Reprod. Biomed. Online 2019, 39, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Popovic, M.; Dheedene, A.; Christodoulou, C.; Taelman, J.; Dhaenens, L.; Van Nieuwerburgh, F.; Deforce, D.; Van den Abbeel, E.; De Sutter, P.; Menten, B.; et al. Chromosomal mosaicism in human blastocysts: The ultimate challenge of preimplantation genetic testing? Hum. Reprod. 2018, 33, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Tsuiko, O.; Zhigalina, D.I.; Jatsenko, T.; Skryabin, N.A.; Kanbekova, O.R.; Artyukhova, V.G.; Svetlakov, A.V.; Teearu, K.; Trosin, A.; Salumets, A.; et al. Karyotype of the blastocoel fluid demonstrates low concordance with both trophectoderm and inner cell mass. Fertil. Steril. 2018, 109, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.T., Jr.; Galliano, D. The challenge of embryonic mosaicism in preimplantation genetic screening. Fertil. Steril. 2016, 105, 1150–1152. [Google Scholar]
- Cuman, C.; Beyer, C.E.; Brodie, D.; Fullston, T.; Lin, J.I.; Willats, E.; Zander-Fox, D.; Mullen, J. Defining the limits of detection for chromosome rearrangements in the preimplantation embryo using next generation sequencing. Hum. Reprod. 2018, 33, 1566–1576. [Google Scholar] [CrossRef]
- Theisen, A.; Shaffer, L.G. Disorders caused by chromosome abnormalities. Appl. Clin. Genet. 2010, 3, 159–174. [Google Scholar]
- Hardy, K.; Handyside, A.H.; Winston, R.M. The human blastocyst: Cell number, death and allocation during late preimplantation development in vitro. Development 1989, 107, 597–604. [Google Scholar]
- Capalbo, A.; Rienzi, L. Mosaicism between trophectoderm and inner cell mass. Fertil. Steril. 2017, 107, 1098–1106. [Google Scholar]
- Huang, J.; Yan, L.; Lu, S.; Zhao, N.; Qiao, J. Re-analysis of aneuploidy blastocysts with an inner cell mass and different regional trophectoderm cells. J. Assist. Reprod. Genet. 2017, 34, 487–493. [Google Scholar]
- Sachdev, N.M.; McCulloh, D.H.; Kramer, Y.; Keefe, D.; Grifo, J.A. The reproducibility of trophectoderm biopsies in euploid, aneuploid, and mosaic embryos using independently verified next-generation sequencing (NGS): A pilot study. J. Assist. Reprod. Genet. 2020, 37, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Lawrenz, B.; El Khatib, I.; Linan, A.; Bayram, A.; Arnanz, A.; Chopra, R.; De Munck, N.; Fatemi, H.M. The clinicians dilemma with mosaicism-an insight from inner cell mass biopsies. Hum. Reprod. 2019, 34, 998–1010. [Google Scholar] [PubMed]
- Victor, A.R.; Griffin, D.K.; Brake, A.J.; Tyndall, J.C.; Murphy, A.E.; Lepkowsky, L.T.; Lal, A.; Zouves, C.G.; Barnes, F.L.; McCoy, R.C.; et al. Assessment of aneuploidy concordance between clinical trophectoderm biopsy and blastocyst. Hum. Reprod. 2019, 34, 181–192. [Google Scholar] [PubMed]
- Navratil, R.; Horak, J.; Hornak, M.; Kubicek, D.; Balcova, M.; Tauwinklova, G.; Travnik, P.; Vesela, K. Concordance of various chromosomal errors among different parts of the embryo and the value of re-biopsy in embryos with segmental aneuploidies. Mol. Hum. Reprod. 2020, 26. [Google Scholar] [CrossRef]
- Ou, Z.; Chen, Z.; Yin, M.; Deng, Y.; Liang, Y.; Wang, W.; Yao, Y.; Sun, L. Re-analysis of whole blastocysts after trophectoderm biopsy indicated chromosome aneuploidy. Hum. Genom. 2020, 14, 3. [Google Scholar]
- Van der Aa, N.; Cheng, J.; Mateiu, L.; Zamani Esteki, M.; Kumar, P.; Dimitriadou, E.; Vanneste, E.; Moreau, Y.; Vermeesch, J.R.; Voet, T. Genome-wide copy number profiling of single cells in S-phase reveals DNA-replication domains. Nucleic Acids Res. 2013, 41, e66. [Google Scholar] [CrossRef]
- Demczuk, A.; Gauthier, M.G.; Veras, I.; Kosiyatrakul, S.; Schildkraut, C.L.; Busslinger, M.; Bechhoefer, J.; Norio, P. Regulation of DNA replication within the immunoglobulin heavy-chain locus during B cell commitment. PLoS Biol. 2012, 10, e1001360. [Google Scholar]
- Dimitriadou, E.; Van der Aa, N.; Cheng, J.; Voet, T.; Vermeesch, J.R. Single cell segmental aneuploidy detection is compromised by S phase. Mol. Cytogenet. 2014, 7, 46. [Google Scholar]
- Ramos, L.; del Rey, J.; Daina, G.; Martinez-Passarell, O.; Rius, M.; Tunon, D.; Campillo, M.; Benet, J.; Navarro, J. Does the S phase have an impact on the accuracy of comparative genomic hybridization profiles in single fibroblasts and human blastomeres? Fertil. Steril. 2014, 101, 488–495. [Google Scholar] [CrossRef]
- Munné, S.; Sultan, K.M.; Weier, H.U.; Grifo, J.A.; Cohen, J.; Rosenwaks, Z. Assessment of numeric abnormalities of X, Y, 18, and 16 chromosomes in preimplantation human embryos before transfer. Am. J. Obs. Gynecol. 1995, 172, 1191–1199. [Google Scholar]
- Rubio, C.; Rodrigo, L.; Perez-Cano, I.; Mercader, A.; Mateu, E.; Buendia, P.; Remohi, J.; Simon, C.; Pellicer, A. FISH screening of aneuploidies in preimplantation embryos to improve IVF outcome. Reprod. Biomed. Online 2005, 11, 497–506. [Google Scholar] [PubMed]
- Verlinsky, Y.; Cieslak, J.; Freidine, M.; Ivakhnenko, V.; Wolf, G.; Kovalinskaya, L.; White, M.; Lifchez, A.; Kaplan, B.; Moise, J.; et al. Pregnancies following pre-conception diagnosis of common aneuploidies by fluorescent in-situ hybridization. Hum. Reprod. 1995, 10, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- Vidal, F.; Gimenez, C.; Rubio, C.; Simon, C.; Pellicer, A.; Santalo, J.; Egozcue, J. FISH preimplantation diagnosis of chromosome aneuploidy in recurrent pregnancy wastage. J. Assist. Reprod. Genet. 1998, 15, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Munné, S.; Wells, D. The cytogenetic constitution of human blastocysts: Insights from comprehensive chromosome screening strategies. Hum. Reprod. Update 2019, 25, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.; Bellver, J.; Rodrigo, L.; Bosch, E.; Mercader, A.; Vidal, C.; De los Santos, M.J.; Giles, J.; Labarta, E.; Domingo, J.; et al. Preimplantation genetic screening using fluorescence in situ hybridization in patients with repetitive implantation failure and advanced maternal age: Two randomized trials. Fertil. Steril. 2013, 99, 1400–1407. [Google Scholar] [PubMed]
- Yang, Z.; Liu, J.; Collins, G.S.; Salem, S.A.; Liu, X.; Lyle, S.S.; Peck, A.C.; Sills, E.S.; Salem, R.D. Selection of single blastocysts for fresh transfer via standard morphology assessment alone and with array CGH for good prognosis IVF patients: Results from a randomized pilot study. Mol. Cytogenet. 2012, 5, 24. [Google Scholar]
- Scott, R.T., Jr.; Ferry, K.; Su, J.; Tao, X.; Scott, K.; Treff, N.R. Comprehensive chromosome screening is highly predictive of the reproductive potential of human embryos: A prospective, blinded, nonselection study. Fertil. Steril. 2012, 97, 870–875. [Google Scholar] [CrossRef]
- Scott, R.T., Jr.; Upham, K.M.; Forman, E.J.; Hong, K.H.; Scott, K.L.; Taylor, D.; Tao, X.; Treff, N.R. Blastocyst biopsy with comprehensive chromosome screening and fresh embryo transfer significantly increases in vitro fertilization implantation and delivery rates: A randomized controlled trial. Fertil. Steril. 2013, 100, 697–703. [Google Scholar]
- Forman, E.J.; Hong, K.H.; Ferry, K.M.; Tao, X.; Taylor, D.; Levy, B.; Treff, N.R.; Scott, R.T., Jr. In vitro fertilization with single euploid blastocyst transfer: A randomized controlled trial. Fertil. Steril. 2013, 100, 100–107. [Google Scholar] [CrossRef]
- Dahdouh, E.M.; Balayla, J.; Garcia-Velasco, J.A. Comprehensive chromosome screening improves embryo selection: A meta-analysis. Fertil. Steril. 2015, 104, 1503–1512. [Google Scholar]
- Chen, M.; Wei, S.; Hu, J.; Quan, S. Can Comprehensive Chromosome Screening Technology Improve IVF/ICSI Outcomes? A Meta-Analysis. PLoS ONE 2015, 10, e0140779. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, L.; Albani, E.; Cesana, A.; Smeraldi, A.; Parini, V.; Fabiani, M.; Poli, M.; Capalbo, A.; Levi-Setti, P.E. Preimplantation Genetic Testing for Aneuploidy Improves Clinical, Gestational, and Neonatal Outcomes in Advanced Maternal Age Patients Without Compromising Cumulative Live-Birth Rate. J. Assist. Reprod. Genet. 2019, 36, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.; Bellver, J.; Rodrigo, L.; Castillon, G.; Guillen, A.; Vidal, C.; Giles, J.; Ferrando, M.; Cabanillas, S.; Remohi, J.; et al. In vitro fertilization with preimplantation genetic diagnosis for aneuploidies in advanced maternal age: A randomized, controlled study. Fertil. Steril. 2017, 107, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Verpoest, W.; Staessen, C.; Bossuyt, P.M.; Goossens, V.; Altarescu, G.; Bonduelle, M.; Devesa, M.; Eldar-Geva, T.; Gianaroli, L.; Griesinger, G.; et al. Preimplantation genetic testing for aneuploidy by microarray analysis of polar bodies in advanced maternal age: A randomized clinical trial. Hum. Reprod. 2018, 33, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Zegers-Hochschild, F.; Adamson, G.D.; Dyer, S.; Racowsky, C.; de Mouzon, J.; Sokol, R.; Rienzi, L.; Sunde, A.; Schmidt, L.; Cooke, I.D.; et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil. Steril. 2017, 108, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Sermon, K.; Capalbo, A.; Cohen, J.; Coonen, E.; De Rycke, M.; De Vos, A.; Delhanty, J.; Fiorentino, F.; Gleicher, N.; Griesinger, G.; et al. The why, the how and the when of PGS 2.0: Current practices and expert opinions of fertility specialists, molecular biologists, and embryologists. Mol. Hum. Reprod. 2016, 22, 845–857. [Google Scholar] [CrossRef]
- Minasi, M.G.; Colasante, A.; Riccio, T.; Ruberti, A.; Casciani, V.; Scarselli, F.; Spinella, F.; Fiorentino, F.; Varricchio, M.T.; Greco, E. Correlation between aneuploidy, standard morphology evaluation and morphokinetic development in 1730 biopsied blastocysts: A consecutive case series study. Hum. Reprod. 2016, 31, 2245–2254. [Google Scholar] [CrossRef]
- Coates, A.; Bankowski, B.J.; Kung, A.; Griffin, D.K.; Munné, S. Differences in pregnancy outcomes in donor egg frozen embryo transfer (FET) cycles following preimplantation genetic screening (PGS): A single center retrospective study. J. Assist. Reprod. Genet. 2017, 34, 71–78. [Google Scholar] [CrossRef]
- Anderson, R.E.; Whitney, J.B.; Schiewe, M.C. Clinical benefits of preimplantation genetic testing for aneuploidy (PGT-A) for all in vitro fertilization treatment cycles. Eur. J. Med. Genet. 2020, 63, 103731. [Google Scholar] [CrossRef]
- Neal, S.A.; Morin, S.J.; Franasiak, J.M.; Goodman, L.R.; Juneau, C.R.; Forman, E.J.; Werner, M.D.; Scott, R.T., Jr. Preimplantation genetic testing for aneuploidy is cost-effective, shortens treatment time, and reduces the risk of failed embryo transfer and clinical miscarriage. Fertil. Steril. 2018, 110, 896–904. [Google Scholar] [CrossRef]
- Somigliana, E.; Busnelli, A.; Paffoni, A.; Vigano, P.; Riccaboni, A.; Rubio, C.; Capalbo, A. Cost-effectiveness of preimplantation genetic testing for aneuploidies. Fertil. Steril. 2019, 111, 1169–1176. [Google Scholar] [PubMed]
- Collins, S.C.; Xu, X.; Mak, W. Cost-effectiveness of preimplantation genetic screening for women older than 37 undergoing in vitro fertilization. J. Assist. Reprod. Genet. 2017, 34, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.; Robinson, W.P. Review: A high capacity of the human placenta for genetic and epigenetic variation: Implications for assessing pregnancy outcome. Placenta 2011, 32, S136–S141. [Google Scholar] [CrossRef] [PubMed]
- Kalousek, D.K.; Barrett, I. Confined placental mosaicism and stillbirth. Pediatr. Pathol. 1994, 14, 151–159. [Google Scholar]
- Viotti, M.; Victor, A.R.; Barnes, F.L.; Zouves, C.G.; Besser, A.G.; Grifo, J.; Cheng, E.H.; Lee, M.S.; Greco, E.; Minasi, M.G.; et al. Mosaic embryos—A comprehensive and powered analysis of clinical outcomes. Fertil. Steril. 2019, 112, e33. [Google Scholar] [CrossRef]
- Grati, F.R. Chromosomal Mosaicism in Human Feto-Placental Development: Implications for Prenatal Diagnosis. J. Clin. Med. 2014, 3, 809–837. [Google Scholar] [CrossRef]
- Kahraman, S.; Cetinkaya, M.; Yuksel, B.; Yesil, M.; Pirkevi Cetinkaya, C. The birth of a baby with mosaicism resulting from a known mosaic embryo transfer: A case report. Hum. Reprod. 2020, 35, 727–733. [Google Scholar] [CrossRef]
- COGEN Position Statement on Chromosomal Mosaicism Detected in Preimplantation Blastocyst Biopsies. Available online: https://ivf-worldwide.com/cogen/oep/publications/cogen-position-statement-on-chromosomal-mosaicism-detected-in-preimplantation-blastocyst-biopsies.html (accessed on 28 May 2020).
- Besser, A.G.; Mounts, E.L. Counselling considerations for chromosomal mosaicism detected by preimplantation genetic screening. Reprod. Biomed. Online 2017, 34, 369–374. [Google Scholar] [CrossRef]
- Besser, A.G.; McCulloh, D.H.; Grifo, J.A. What are patients doing with their mosaic embryos? Decision making after genetic counseling. Fertil. Steril. 2019, 111, 132–137. [Google Scholar] [CrossRef]
- Diez-Juan, A.; Rubio, C.; Marin, C.; Martinez, S.; Al-Asmar, N.; Riboldi, M.; Diaz-Gimeno, P.; Valbuena, D.; Simon, C. Mitochondrial DNA content as a viability score in human euploid embryos: Less is better. Fertil. Steril. 2015, 104, 534–541. [Google Scholar] [CrossRef]
- Fragouli, E.; Spath, K.; Alfarawati, S.; Kaper, F.; Craig, A.; Michel, C.E.; Kokocinski, F.; Cohen, J.; Munné, S.; Wells, D. Altered levels of mitochondrial DNA are associated with female age, aneuploidy, and provide an independent measure of embryonic implantation potential. PLoS Genet. 2015, 11, e1005241. [Google Scholar]
- Ravichandran, K.; McCaffrey, C.; Grifo, J.; Morales, A.; Perloe, M.; Munné, S.; Wells, D.; Fragouli, E. Mitochondrial DNA quantification as a tool for embryo viability assessment: Retrospective analysis of data from single euploid blastocyst transfers. Hum. Reprod. 2017, 32, 1282–1292. [Google Scholar] [CrossRef]
- Fragouli, E.; McCaffrey, C.; Ravichandran, K.; Spath, K.; Grifo, J.A.; Munné, S.; Wells, D. Clinical implications of mitochondrial DNA quantification on pregnancy outcomes: A blinded prospective non-selection study. Hum. Reprod. 2017, 32, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Lledo, B.; Ortiz, J.A.; Morales, R.; Garcia-Hernandez, E.; Ten, J.; Bernabeu, A.; Llacer, J.; Bernabeu, R. Comprehensive mitochondrial DNA analysis and IVF outcome. Hum. Reprod. Open 2018, 2018, hoy023. [Google Scholar]
- Klimczak, A.M.; Pacheco, L.E.; Lewis, K.E.; Massahi, N.; Richards, J.P.; Kearns, W.G.; Saad, A.F.; Crochet, J.R. Embryonal mitochondrial DNA: Relationship to embryo quality and transfer outcomes. J. Assist. Reprod. Genet. 2018, 35, 871–877. [Google Scholar]
- Treff, N.R.; Zhan, Y.; Tao, X.; Olcha, M.; Han, M.; Rajchel, J.; Morrison, L.; Morin, S.J.; Scott, R.T., Jr. Levels of trophectoderm mitochondrial DNA do not predict the reproductive potential of sibling embryos. Hum. Reprod. 2017, 32, 954–962. [Google Scholar] [PubMed]
- Victor, A.; Griffin, D.; Dardner, K.G.; Brake, A.; Zouves, C.; Barnes, F.; Viotti, M. Births from embryos with highly elevated levels of mitochondrial DNA. Reprod. Biomed. Online 2019, 39, 403–412. [Google Scholar] [CrossRef]
- Victor, A.R.; Brake, A.J.; Tyndall, J.C.; Griffin, D.K.; Zouves, C.G.; Barnes, F.L.; Viotti, M. Accurate quantitation of mitochondrial DNA reveals uniform levels in human blastocysts irrespective of ploidy, age, or implantation potential. Fertil. Steril. 2017, 107, 34–42. [Google Scholar] [CrossRef]
- Lee, Y.X.; Chen, C.H.; Lin, S.Y.; Lin, Y.H.; Tzeng, C.R. Adjusted mitochondrial DNA quantification in human embryos may not be applicable as a biomarker of implantation potential. J. Assist. Reprod. Genet. 2019, 36, 1855–1865. [Google Scholar]
- de Los Santos, M.J.; Diez Juan, A.; Mifsud, A.; Mercader, A.; Meseguer, M.; Rubio, C.; Pellicer, A. Variables associated with mitochondrial copy number in human blastocysts: What can we learn from trophectoderm biopsies? Fertil. Steril. 2018, 109, 110–117. [Google Scholar] [CrossRef]
- Piko, L.; Taylor, K.D. Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Dev. Biol. 1987, 123, 364–374. [Google Scholar] [PubMed]
- Hashimoto, S.; Morimoto, N.; Yamanaka, M.; Matsumoto, H.; Yamochi, T.; Goto, H.; Inoue, M.; Nakaoka, Y.; Shibahara, H.; Morimoto, Y. Quantitative and qualitative changes of mitochondria in human preimplantation embryos. J. Assist. Reprod. Genet. 2017, 34, 573–580. [Google Scholar]
- St John, J. The control of mtDNA replication during differentiation and development. Biochim. Biophys. Acta 2014, 1840, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. FAST-SeqS: A simple and efficient method for the detection of aneuploidy by massively parallel sequencing. PLoS ONE 2012, 7, e41162. [Google Scholar] [CrossRef] [PubMed]
- Marin, D.; Zimmerman, R.; Tao, X.; Zhan, Y.; Scott, R.T., Jr.; Treff, N.R. Validation of a targeted next generation sequencing-based comprehensive chromosome screening platform for detection of triploidy in human blastocysts. Reprod. Biomed. Online 2018, 36, 388–395. [Google Scholar] [PubMed]
- Masset, H.; Zamani Esteki, M.; Dimitriadou, E.; Dreesen, J.; Debrock, S.; Derhaag, J.; Derks, K.; Destouni, A.; Drusedau, M.; Meekels, J.; et al. Multi-centre evaluation of a comprehensive preimplantation genetic test through haplotyping-by-sequencing. Hum. Reprod. 2019, 34, 1608–1619. [Google Scholar] [CrossRef]
- Zamani Esteki, M.; Dimitriadou, E.; Mateiu, L.; Melotte, C.; Van der Aa, N.; Kumar, P.; Das, R.; Theunis, K.; Cheng, J.; Legius, E.; et al. Concurrent whole-genome haplotyping and copy-number profiling of single cells. Am. J. Hum. Genet. 2015, 96, 894–912. [Google Scholar] [CrossRef]
- Zimmerman, R.S.; Jalas, C.; Tao, X.; Fedick, A.M.; Kim, J.G.; Pepe, R.J.; Northrop, L.E.; Scott, R.T., Jr.; Treff, N.R. Development and validation of concurrent preimplantation genetic diagnosis for single gene disorders and comprehensive chromosomal aneuploidy screening without whole genome amplification. Fertil. Steril. 2016, 105, 286–294. [Google Scholar] [CrossRef]
- Kimura, Y.; Laliberte, J.; Kamberov, E.; Viotti, M.; Victor, A.R.; Brake, A.J.; Zouves, C.G.; Barnes, F.L.; Farmer, A. Novel approach enabling the simultaneous detection of snv and cnv for pgt-m and pgt-a using a single-tube assay. Reprod. Biomed. Online 2019, 39, E16–E17. [Google Scholar]
- Del Rey, J.; Vidal, F.; Ramirez, L.; Borras, N.; Corrales, I.; Garcia, I.; Martinez-Pasarell, O.; Fernandez, S.F.; Garcia-Cruz, R.; Pujol, A.; et al. Novel Double Factor PGT strategy analyzing blastocyst stage embryos in a single NGS procedure. PLoS ONE 2018, 13, e0205692. [Google Scholar]
- Alcaraz Mas, L.A.; Pérez, C.; González-Reig, S.; Brígido, P.; Amorós, D.; Penacho, V.; Blanca, H. Pgd-seq: Validation of a novel solution for pgt-m and pgt-sr based on target enrichment. Reprod. Biomed. Online 2019, 39, E62. [Google Scholar] [CrossRef]
- Handyside, A.H.; Harton, G.L.; Mariani, B.; Thornhill, A.R.; Affara, N.; Shaw, M.A.; Griffin, D.K. Karyomapping: A universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med. Genet. 2010, 47, 651–658. [Google Scholar] [PubMed]
- Natesan, S.A.; Handyside, A.H.; Thornhill, A.R.; Ottolini, C.S.; Sage, K.; Summers, M.C.; Konstantinidis, M.; Wells, D.; Griffin, D.K. Live birth after PGD with confirmation by a comprehensive approach (karyomapping) for simultaneous detection of monogenic and chromosomal disorders. Reprod. Biomed. Online 2014, 29, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, A.R.; Handyside, A.H.; Ottolini, C.; Natesan, S.A.; Taylor, J.; Sage, K.; Harton, G.; Cliffe, K.; Affara, N.; Konstantinidis, M.; et al. Karyomapping-a comprehensive means of simultaneous monogenic and cytogenetic PGD: Comparison with standard approaches in real time for Marfan syndrome. J. Assist. Reprod. Genet. 2015, 32, 347–356. [Google Scholar] [CrossRef]
- Treff, N.R.; Eccles, J.; Lello, L.; Bechor, E.; Hsu, J.; Plunkett, K.; Zimmerman, R.; Rana, B.; Samoilenko, A.; Hsu, S.; et al. Utility and First Clinical Application of Screening Embryos for Polygenic Disease Risk Reduction. Front. Endocrinol. 2019, 10, 845. [Google Scholar] [CrossRef]
- Treff, N.R.; Zimmerman, R.; Bechor, E.; Hsu, J.; Rana, B.; Jensen, J.; Li, J.; Samoilenko, A.; Mowrey, W.; Van Alstine, J.; et al. Validation of concurrent preimplantation genetic testing for polygenic and monogenic disorders, structural rearrangements, and whole and segmental chromosome aneuploidy with a single universal platform. Eur. J. Med. Genet. 2019, 62, 103647. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Wei, S.; Weiss, Z.R.; Gaur, P.; Forman, E.; Williams, Z. Rapid preimplantation genetic screening using a handheld, nanopore-based DNA sequencer. Fertil. Steril. 2018, 110, 910–916. [Google Scholar] [CrossRef]
- Kumar, A.; Ryan, A.; Kitzman, J.O.; Wemmer, N.; Snyder, M.W.; Sigurjonsson, S.; Lee, C.; Banjevic, M.; Zarutskie, P.W.; Lewis, A.P.; et al. Whole genome prediction for preimplantation genetic diagnosis. Genome Med. 2015, 7, 35. [Google Scholar] [CrossRef][Green Version]
- Peters, B.A.; Kermani, B.G.; Alferov, O.; Agarwal, M.R.; McElwain, M.A.; Gulbahce, N.; Hayden, D.M.; Tang, Y.T.; Zhang, R.Y.; Tearle, R.; et al. Detection and phasing of single base de novo mutations in biopsies from human in vitro fertilized embryos by advanced whole-genome sequencing. Genome Res. 2015, 25, 426–434. [Google Scholar] [CrossRef]
- Murphy, N.M.; Samarasekera, T.S.; Macaskill, L.; Mullen, J.; Rombauts, L.J.F. Genome sequencing of human in vitro fertilisation embryos for pathogenic variation screening. Sci. Rep. 2020, 10, 3795. [Google Scholar] [CrossRef] [PubMed]
- Stigliani, S.; Persico, L.; Lagazio, C.; Anserini, P.; Venturini, P.L.; Scaruffi, P. Mitochondrial DNA in Day 3 embryo culture medium is a novel, non-invasive biomarker of blastocyst potential and implantation outcome. Mol. Hum. Reprod. 2014, 20, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Palini, S.; Galluzzi, L.; De Stefani, S.; Bianchi, M.; Wells, D.; Magnani, M.; Bulletti, C. Genomic DNA in human blastocoele fluid. Reprod. Biomed. Online 2013, 26, 603–610. [Google Scholar] [CrossRef]
- Gianaroli, L.; Magli, M.C.; Pomante, A.; Crivello, A.M.; Cafueri, G.; Valerio, M.; Ferraretti, A.P. Blastocentesis: A source of DNA for preimplantation genetic testing. Results from a pilot study. Fertil. Steril. 2014, 102, 1692–1699. [Google Scholar] [CrossRef]
- Magli, M.C.; Albanese, C.; Crippa, A.; Tabanelli, C.; Ferraretti, A.P.; Gianaroli, L. Deoxyribonucleic acid detection in blastocoelic fluid: A new predictor of embryo ploidy and viable pregnancy. Fertil. Steril. 2019, 111, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Magli, M.C.; Pomante, A.; Cafueri, G.; Valerio, M.; Crippa, A.; Ferraretti, A.P.; Gianaroli, L. Preimplantation genetic testing: Polar bodies, blastomeres, trophectoderm cells, or blastocoelic fluid? Fertil. Steril. 2016, 105, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Tobler, K.J.; Zhao, Y.; Ross, R.; Benner, A.T.; Xu, X.; Du, L.; Broman, K.; Thrift, K.; Brezina, P.R.; Kearns, W.G. Blastocoel fluid from differentiated blastocysts harbors embryonic genomic material capable of a whole-genome deoxyribonucleic acid amplification and comprehensive chromosome microarray analysis. Fertil. Steril. 2015, 104, 418–425. [Google Scholar] [CrossRef]
- Capalbo, A.; Romanelli, V.; Patassini, C.; Poli, M.; Girardi, L.; Giancani, A.; Stoppa, M.; Cimadomo, D.; Ubaldi, F.M.; Rienzi, L. Diagnostic efficacy of blastocoel fluid and spent media as sources of DNA for preimplantation genetic testing in standard clinical conditions. Fertil. Steril. 2018, 110, 870–879. [Google Scholar] [CrossRef]
- Hammond, E.R.; McGillivray, B.C.; Wicker, S.M.; Peek, J.C.; Shelling, A.N.; Stone, P.; Chamley, L.W.; Cree, L.M. Characterizing nuclear and mitochondrial DNA in spent embryo culture media: Genetic contamination identified. Fertil. Steril. 2017, 107, 220–228. [Google Scholar] [CrossRef]
- Vera-Rodriguez, M.; Diez-Juan, A.; Jimenez-Almazan, J.; Martinez, S.; Navarro, R.; Peinado, V.; Mercader, A.; Meseguer, M.; Blesa, D.; Moreno, I.; et al. Origin and composition of cell-free DNA in spent medium from human embryo culture during preimplantation development. Hum. Reprod. 2018, 33, 745–756. [Google Scholar] [CrossRef]
- Shamonki, M.I.; Jin, H.; Haimowitz, Z.; Liu, L. Proof of concept: Preimplantation genetic screening without embryo biopsy through analysis of cell-free DNA in spent embryo culture media. Fertil. Steril. 2016, 106, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Leaver, M.; Wells, D. Non-invasive preimplantation genetic testing (niPGT): The next revolution in reproductive genetics? Hum. Reprod. Update 2020, 26, 16–42. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fang, R.; Chen, L.; Chen, D.; Xiao, J.P.; Yang, W.; Wang, H.; Song, X.; Ma, T.; Bo, S.; et al. Noninvasive chromosome screening of human embryos by genome sequencing of embryo culture medium for in vitro fertilization. Proc. Natl. Acad. Sci. USA 2016, 113, 11907–11912. [Google Scholar] [PubMed]
- Feichtinger, M.; Vaccari, E.; Carli, L.; Wallner, E.; Madel, U.; Figl, K.; Palini, S.; Feichtinger, W. Non-invasive preimplantation genetic screening using array comparative genomic hybridization on spent culture media: A proof-of-concept pilot study. Reprod. Biomed. Online 2017, 34, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.R.; Arrach, N.; Rhodes-Long, K.; Ahmady, A.; Ingles, S.; Chung, K.; Bendikson, K.A.; Paulson, R.J.; McGinnis, L.K. Pushing the limits of detection: Investigation of cell-free DNA for aneuploidy screening in embryos. Fertil. Steril. 2018, 110, 467–475. [Google Scholar]
- Huang, L.; Bogale, B.; Tang, Y.; Lu, S.; Xie, X.S.; Racowsky, C. Noninvasive preimplantation genetic testing for aneuploidy in spent medium may be more reliable than trophectoderm biopsy. Proc. Natl. Acad. Sci. USA 2019, 116, 14105–14112. [Google Scholar] [CrossRef]
- Yeung, Q.S.Y.; Zhang, Y.X.; Chung, J.P.W.; Lui, W.T.; Kwok, Y.K.Y.; Gui, B.; Kong, G.W.S.; Cao, Y.; Li, T.C.; Choy, K.W. A prospective study of non-invasive preimplantation genetic testing for aneuploidies (NiPGT-A) using next-generation sequencing (NGS) on spent culture media (SCM). J. Assist. Reprod. Genet. 2019, 36, 1609–1621. [Google Scholar] [CrossRef]
- Liu, W.; Liu, J.; Du, H.; Ling, J.; Sun, X.; Chen, D. Non-invasive pre-implantation aneuploidy screening and diagnosis of beta thalassemia IVSII654 mutation using spent embryo culture medium. Ann. Med. 2017, 49, 319–328. [Google Scholar] [CrossRef]
- Fang, R.; Yang, W.; Zhao, X.; Xiong, F.; Guo, C.; Xiao, J.; Chen, L.; Song, X.; Wang, H.; Chen, J.; et al. Chromosome screening using culture medium of embryos fertilised in vitro: A pilot clinical study. J. Transl. Med. 2019, 17, 73. [Google Scholar]
- Rubio, C.; Rienzi, L.; Navarro-Sanchez, L.; Cimadomo, D.; Garcia-Pascual, C.M.; Albricci, L.; Soscia, D.; Valbuena, D.; Capalbo, A.; Ubaldi, F.; et al. Embryonic cell-free DNA versus trophectoderm biopsy for aneuploidy testing: Concordance rate and clinical implications. Fertil. Steril. 2019, 112, 510–519. [Google Scholar] [CrossRef]
- Li, P.; Song, Z.; Yao, Y.; Huang, T.; Mao, R.; Huang, J.; Ma, Y.; Dong, X.; Huang, W.; Huang, J.; et al. Preimplantation Genetic Screening with Spent Culture Medium/Blastocoel Fluid for in Vitro Fertilization. Sci. Rep. 2018, 8, 9275. [Google Scholar] [PubMed]
- Kuznyetsov, V.; Madjunkova, S.; Antes, R.; Abramov, R.; Motamedi, G.; Ibarrientos, Z.; Librach, C. Evaluation of a novel non-invasive preimplantation genetic screening approach. PLoS ONE 2018, 13, e0197262. [Google Scholar]
- Jiao, J.; Shi, B.; Sagnelli, M.; Yang, D.; Yao, Y.; Li, W.; Shao, L.; Lu, S.; Li, D.; Wang, X. Minimally invasive preimplantation genetic testing using blastocyst culture medium. Hum. Reprod. 2019, 34, 1369–1379. [Google Scholar]
- Kuznyetsov, V.; Madjunkova, S.; Abramov, R.; Antes, R.; Ibarrientos, Z.; Motamedi, G.; Zaman, A.; Kuznyetsova, I.; Librach, C.L. Minimally Invasive Cell-Free Human Embryo Aneuploidy Testing (miPGT-A) Utilizing Combined Spent Embryo Culture Medium and Blastocoel Fluid-Towards Development of a Clinical Assay. Sci. Rep. 2020, 10, 7244. [Google Scholar] [CrossRef] [PubMed]
- Stigliani, S.; Orlando, G.; Massarotti, C.; Casciano, I.; Bovis, F.; Anserini, P.; Ubaldi, F.M.; Remorgida, V.; Rienzi, L.; Scaruffi, P. Non-invasive mitochondrial DNA quantification on Day 3 predicts blastocyst development: A prospective, blinded, multi-centric study. Mol. Hum. Reprod. 2019, 25, 527–537. [Google Scholar] [PubMed]
- Theobald, R.; SenGupta, S.; Harper, J. The status of preimplantation genetic testing in the UK and USA. Hum. Reprod. 2020, 35, 986–998. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viotti, M. Preimplantation Genetic Testing for Chromosomal Abnormalities: Aneuploidy, Mosaicism, and Structural Rearrangements. Genes 2020, 11, 602. https://doi.org/10.3390/genes11060602
Viotti M. Preimplantation Genetic Testing for Chromosomal Abnormalities: Aneuploidy, Mosaicism, and Structural Rearrangements. Genes. 2020; 11(6):602. https://doi.org/10.3390/genes11060602
Chicago/Turabian StyleViotti, Manuel. 2020. "Preimplantation Genetic Testing for Chromosomal Abnormalities: Aneuploidy, Mosaicism, and Structural Rearrangements" Genes 11, no. 6: 602. https://doi.org/10.3390/genes11060602
APA StyleViotti, M. (2020). Preimplantation Genetic Testing for Chromosomal Abnormalities: Aneuploidy, Mosaicism, and Structural Rearrangements. Genes, 11(6), 602. https://doi.org/10.3390/genes11060602