Genome-Wide Patterns of Homozygosity and Relevant Characterizations on the Population Structure in Piétrain Pigs


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. Linkage Disequilibrium and Effective Population Size
2.3. Identification of Runs of Homozygosity
2.4. Inbreeding Coefficient
2.5. Functional Annotation for Core Regions of ROH
3. Results
3.1. Characteristics of Genotypic Data
3.2. The Patterns of Linkage Disequilibrium and Past Effective Population Size
3.3. Identification and Classification of ROH
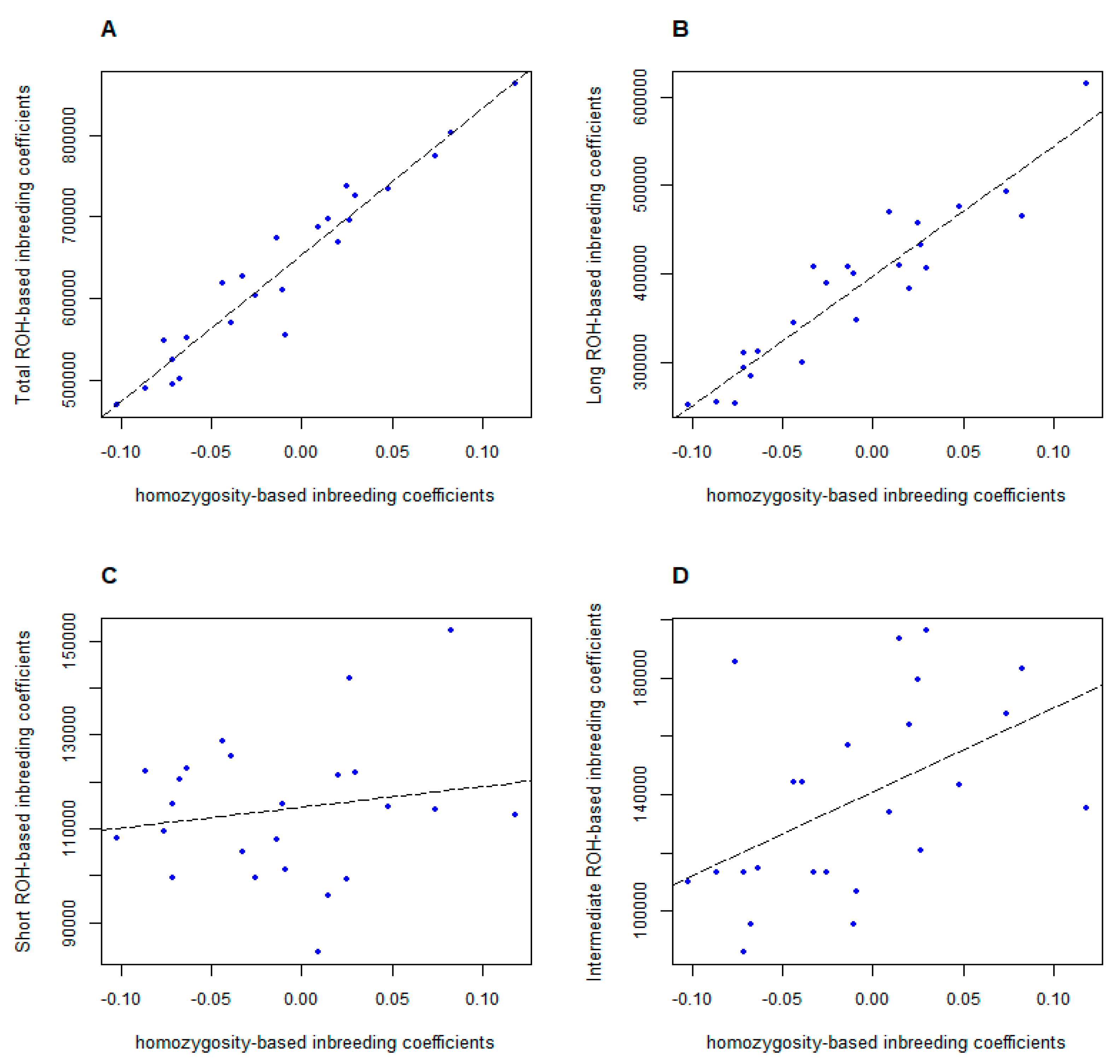
3.4. Comparison of Different Inbreeding Coefficients
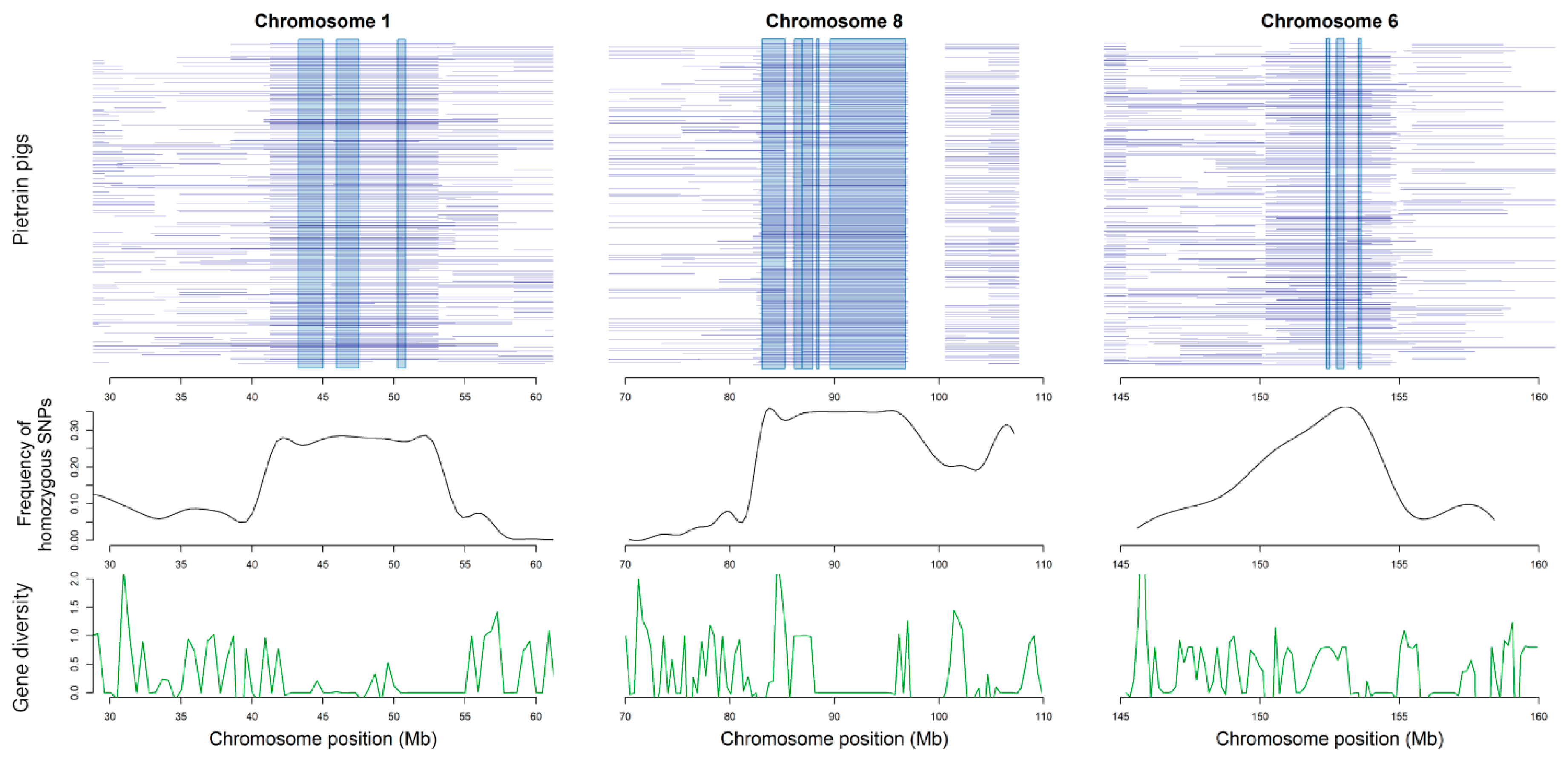
3.5. Functional Annotation for Core Regions of ROH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gorssen, W.; Meyermans, R.; Buys, N.; Janssens, S. SNP genotypes reveal breed substructure, selection signatures and highly inbred regions in Pietrain pigs. Anim. Genet. 2020, 51, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Porter, V. Pigs: A Handbook to the Breeds of the World; Comstock Publishing Associates: Ithaca, NY, USA, 1993; Volume 1, pp. 129–130. [Google Scholar]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Ciani, E.; Sardina, M.T.; Sottile, G.; Pilla, F.; Portolano, B. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef]
- Bosse, M.; Megens, H.J.; Madsen, O.; Paudel, Y.; Frantz, L.A.; Schook, L.B.; Crooijmans, R.P.; Groenen, M.A. Regions of homozygosity in the porcine genome: Consequence of demography and the recombination landscape. PLoS Genet. 2012, 8, e1003100. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef]
- Szmatoła, T.; Gurgul, A.; Ropka-Molik, K.; Jasielczuk, I.; Zabek, T.; Bugno, M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest. Sci. 2016, 188, 72–80. [Google Scholar] [CrossRef]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in U.S. Holstein cattle. PLoS ONE 2013, 8, e80813. [Google Scholar] [CrossRef] [PubMed]
- Pryce, J.E.; Haile-Mariam, M.; Goddard, M.E.; Hayes, B.J. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle. Genet. Sel. Evol. 2014, 46, 71. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef]
- Balmain, A. Cancer: New-age tumour suppressors. Nature 2002, 417, 235–237. [Google Scholar] [CrossRef]
- Ismail, J.; Jafar, T.H.; Jafary, F.H.; White, F.; Faruqui, A.M.; Chaturvedi, N. Risk factors for non-fatal myocardial infarction in young South Asian adults. Heart 2004, 90, 259–263. [Google Scholar] [CrossRef]
- Garrod, A.E. The Incidence of Alkaptonuria: A Study in Chemical Individuality. Lancet 1902, 160, 1616–1620. [Google Scholar] [CrossRef]
- Woods, C.G.; Cox, J.; Springell, K.; Hampshire, D.J.; Mohamed, M.D.; McKibbin, M.; Stern, R.; Raymond, F.L.; Sandford, R.; Malik Sharif, S.; et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006, 78, 889–896. [Google Scholar] [CrossRef]
- Wright, S. Wright s. Size of population and breeding structure in relation to evolution. Science 1938, 87, 430–431. [Google Scholar]
- Sved, J.A. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- Hayes, B.J.; Visscher, P.M.; McPartlan, H.C.; Goddard, M.E. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003, 13, 635–643. [Google Scholar] [CrossRef]
- Tenesa, A.; Navarro, P.; Hayes, B.J.; Duffy, D.L.; Clarke, G.M.; Goddard, M.E.; Visscher, P.M. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007, 17, 520–526. [Google Scholar] [CrossRef]
- Villa-Angulo, R.; Matukumalli, L.K.; Gill, C.A.; Choi, J.; Van Tassell, C.P.; Grefenstette, J.J. High-resolution haplotype block structure in the cattle genome. BMC Genet. 2009, 10, 19. [Google Scholar] [CrossRef]
- Corbin, L.J.; Blott, S.C.; Swinburne, J.E.; Vaudin, M.; Bishop, S.C.; Woolliams, J.A. Linkage disequilibrium and historical effective population size in the Thoroughbred horse. Anim. Genet. 2010, 41, 8–15. [Google Scholar] [CrossRef]
- Shin, D.; Won, K.H.; Kim, S.H.; Kim, Y.M. Extent of linkage disequilibrium and effective population size of Korean Yorkshire swine. Asian-Australas. J. Anim. Sci. 2018, 31, 1843–1851. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Qanbari, S.; Hansen, M.; Weigend, S.; Preisinger, R.; Simianer, H. Linkage disequilibrium reveals different demographic history in egg laying chickens. BMC Genet. 2010, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Uimari, P.; Tapio, M. Extent of linkage disequilibrium and effective population size in Finnish Landrace and Finnish Yorkshire pig breeds. J. Anim. Sci. 2011, 89, 609–614. [Google Scholar] [CrossRef]
- Srisai, D.; Yin, T.C.; Lee, A.A.; Rouault, A.A.J.; Pearson, N.A.; Grobe, J.L.; Sebag, J.A. MRAP2 regulates ghrelin receptor signaling and hunger sensing. Nat. Commun. 2017, 8, 713. [Google Scholar] [CrossRef]
- Shiba, T.; Kakuda, S.; Ishiguro, M.; Morita, I.; Oka, S.; Kawasaki, T.; Wakatsuki, S.; Kato, R. Crystal structure of GlcAT-S, a human glucuronyltransferase, involved in the biosynthesis of the HNK-1 carbohydrate epitope. Proteins 2006, 65, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Gailus, S.; Miousse, I.R.; Suormala, T.; Sagne, C.; Toliat, M.R.; Nurnberg, G.; Wittkampf, T.; Buers, I.; Sharifi, A.; et al. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet. 2009, 41, 234–239. [Google Scholar] [CrossRef]
- Nolte, I.M.; Wallace, C.; Newhouse, S.J.; Waggott, D.; Fu, J.; Soranzo, N.; Gwilliam, R.; Deloukas, P.; Savelieva, I.; Zheng, D.; et al. Common genetic variation near the phospholamban gene is associated with cardiac repolarisation: Meta-analysis of three genome-wide association studies. PLoS ONE 2009, 4, e6138. [Google Scholar] [CrossRef]
- Schmoker, A.M.; Weinert, J.L.; Kellett, K.J.; Johnson, H.E.; Joy, R.M.; Weir, M.E.; Ebert, A.M.; Ballif, B.A. Dynamic multi-site phosphorylation by Fyn and Abl drives the interaction between CRKL and the novel scaffolding receptors DCBLD1 and DCBLD2. Biochem. J. 2017, 474, 3963–3984. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal degradation of YME1L and OMA1 adapts mitochondrial proteolytic activity during stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef]
- Van Lith, M.; Karala, A.R.; Bown, D.; Gatehouse, J.A.; Ruddock, L.W.; Saunders, P.T.; Benham, A.M. A developmentally regulated chaperone complex for the endoplasmic reticulum of male haploid germ cells. Mol. Biol. Cell 2007, 18, 2795–2804. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frazer, K.A.; Ballinger, D.G.; Cox, D.R.; Hinds, D.A.; Stuve, L.L.; Gibbs, R.A.; Belmont, J.W.; Boudreau, A.; Hardenbol, P.; Leal, S.M.; et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Stratz, P.; Wimmers, K.; Meuwissen, T.H.; Bennewitz, J. Investigations on the pattern of linkage disequilibrium and selection signatures in the genomes of German Pietrain pigs. J. Anim. Breed. Genet. 2014, 131, 473–482. [Google Scholar] [CrossRef]
- Corbin, L.J.; Liu, A.Y.; Bishop, S.C.; Woolliams, J.A. Estimation of historical effective population size using linkage disequilibria with marker data. J. Anim. Breed. Genet. 2012, 129, 257–270. [Google Scholar] [CrossRef]
- Sölkner, J.; Ferenčaković, M.; Schwarzenbacher, H.; Gredler, B.; Fürst, C.; Čurik, I. Genomic Metrics of Individual Autozygosity, Applied to a Cattle Population. In Proceedings of the 61st Annual Conference of the European Association for Animal Production, Heraklion, Greece, 23–27 August 2010. Session code G38.03. [Google Scholar]
- Zavarez, L.B.; Utsunomiya, Y.T.; Carmo, A.S.; Neves, H.H.; Carvalheiro, R.; Ferencakovic, M.; Perez O’Brien, A.M.; Curik, I.; Cole, J.B.; Van Tassell, C.P.; et al. Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes. Front. Genet. 2015, 6, 5. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.; Ventura, R.V.; Baldi, F.; da Silva, M. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef]
- Broman, K.W.; Weber, J.L. Long homozygous chromosomal segments in reference families from the Centre d’Etude du Polymorphisme Humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L.; et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.H.; Wang, B.H.; Xia, W.; Mo, X.B.; Wu, L.F.; Zhu, X.W.; He, P.; Xie, F.F.; Lu, X.; Deng, F.Y.; et al. The distribution and functional relevance analysis of runs of homozygosity (ROHs) in Chinese Han female population. Mol. Genet. Genom. 2018, 293, 197–206. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr. | cM/Mb | linkage Disequilibrium (r2) of Different Interval Distance (Kb) | Estimated Ne in n Generations Ago | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 | 200 | 500 | 1000 | 2000 | 5000 | 500 | 250 | 100 | 50 | 25 | 10 | ||
| 1 | 0.5365 | 0.4472 | 0.3956 | 0.3188 | 0.2413 | 0.1666 | 0.0932 | 576.1 | 356.0 | 199.1 | 146.5 | 116.5 | 90.7 |
| 2 | 0.8691 | 0.4464 | 0.3976 | 0.3210 | 0.2474 | 0.1721 | 0.0977 | 356.8 | 218.0 | 121.7 | 87.5 | 69.2 | 53.1 |
| 3 | 0.9723 | 0.4486 | 0.4018 | 0.3248 | 0.2493 | 0.1752 | 0.1000 | 316.1 | 191.4 | 106.9 | 77.4 | 60.5 | 46.3 |
| 4 | 1.0000 | 0.4482 | 0.3980 | 0.3229 | 0.2488 | 0.1753 | 0.1004 | 307.8 | 189.1 | 104.8 | 75.5 | 58.8 | 44.8 |
| 5 | 1.4524 | 0.4518 | 0.4019 | 0.3264 | 0.2517 | 0.1766 | 0.1014 | 208.9 | 128.1 | 71.0 | 51.2 | 40.1 | 30.5 |
| 6 | 0.9690 | 0.4477 | 0.3960 | 0.3204 | 0.2457 | 0.1718 | 0.0989 | 318.3 | 196.7 | 109.4 | 79.2 | 62.2 | 47.0 |
| 7 | 1.2869 | 0.4495 | 0.4018 | 0.3253 | 0.2495 | 0.1749 | 0.1006 | 238.0 | 144.6 | 80.6 | 58.4 | 45.8 | 34.7 |
| 8 | 0.9403 | 0.4480 | 0.3999 | 0.3239 | 0.2475 | 0.1731 | 0.0991 | 327.7 | 199.5 | 111.0 | 80.9 | 63.5 | 48.3 |
| 9 | 0.9893 | 0.4462 | 0.3989 | 0.3238 | 0.2476 | 0.1747 | 0.1006 | 313.6 | 190.4 | 105.5 | 76.8 | 59.7 | 45.2 |
| 10 | 1.8551 | 0.4508 | 0.4012 | 0.3249 | 0.2490 | 0.1755 | 0.1010 | 164.2 | 100.6 | 56.0 | 40.6 | 31.7 | 24.0 |
| 11 | 1.0747 | 0.4504 | 0.4015 | 0.3248 | 0.2494 | 0.1746 | 0.0999 | 283.9 | 173.4 | 96.7 | 70.0 | 55.0 | 41.9 |
| 12 | 1.8242 | 0.4482 | 0.3994 | 0.3229 | 0.2486 | 0.1744 | 0.1006 | 168.7 | 103.1 | 57.5 | 41.4 | 32.4 | 24.5 |
| 13 | 0.6067 | 0.4505 | 0.4031 | 0.3255 | 0.2490 | 0.1742 | 0.0991 | 502.6 | 305.1 | 170.8 | 124.3 | 97.6 | 74.9 |
| 14 | 0.7331 | 0.4515 | 0.3999 | 0.3217 | 0.2465 | 0.1714 | 0.0982 | 414.3 | 255.9 | 143.8 | 104.2 | 82.4 | 62.6 |
| 15 | 0.9021 | 0.4470 | 0.4006 | 0.3242 | 0.2485 | 0.1747 | 0.1005 | 342.9 | 207.4 | 115.5 | 83.8 | 65.5 | 49.6 |
| 16 | 1.1650 | 0.4498 | 0.4015 | 0.3247 | 0.2503 | 0.1758 | 0.1003 | 262.5 | 160.0 | 89.3 | 64.3 | 50.3 | 38.5 |
| 17 | 1.5397 | 0.4488 | 0.3997 | 0.3234 | 0.2487 | 0.1740 | 0.0998 | 199.4 | 121.9 | 67.9 | 49.0 | 38.5 | 29.3 |
| 18 | 1.1859 | 0.4476 | 0.3980 | 0.3222 | 0.2476 | 0.1733 | 0.1003 | 260.1 | 159.5 | 88.7 | 64.1 | 50.3 | 37.8 |
| Total | 1.1057 | 0.4488 | 0.3998 | 0.3234 | 0.2481 | 0.1738 | 0.0995 | 309.0 | 188.9 | 105.4 | 76.4 | 60.0 | 45.8 |
| Category | Trait | QTL ID |
|---|---|---|
| Meat and Carcass | Anatomy | QTL:28022,QTL:8699,QTL:154422,QTL:29582,QTL:10245 |
| Fat composition | QTL:31377,QTL:31364,QTL:31383,QTL:32083,QTL:18547,QTL:101788,QTL:101825,QTL:31356,QTL:31374,QTL:101552,QTL:18546,QTL:31372,QTL:31361,QTL:31368,QTL:18548,QTL:101865,QTL:31360,QTL:6435,QTL:31369,QTL:18549,QTL:101898,QTL:31370 | |
| fatness | QTL:22293,QTL:139226 | |
| Meat color | QTL:36140,QTL:36138 | |
| pH | QTL:23051,QTL:18667,QTL:5871 | |
| texture | QTL:23050,QTL:7819,QTL:7885,QTL:3864,QTL:23052 | |
| Chemistry | QTL:9042 | |
| Health | blood parameters | QTL:29669,QTL:29665,QTL:29663,QTL:29668,QTL:29670,QTL:29666,QTL:6549,QTL:6318,QTL:29667 |
| Immune capacity | QTL:127811 | |
| Exterior | QTL:124729,QTL:17665,QTL:32743,QTL:16383,QTL:125736,QTL:64738,QTL:64740,QTL:64686 | |
| Production | feed conversion | QTL:27977 |
| feed intake | QTL:31324 | |
| Growth | QTL:22971 | |
| Reproduction | Litter traits | QTL:31861,QTL:122507,QTL:64741,QTL:18327 |
| Reproductive organs | QTL:18371,QTL:126623,QTL:126620 | |
| Reproductive traits | QTL:7662,QTL:18326,QTL:18329,QTL:18328,QTL:18325 |
| Chr. | Position (Kb) | Length (Kb) | ROH Mapping Frequency1 | Genes | Gene Function |
|---|---|---|---|---|---|
| 1 | 53078–53113; | 35.052; | 76.38%; | MRAP2 | regulate the energy homeostasis machinery [32] |
| 1 | 50245–50798; | 553.125; | 73.14%; | B3GAT2 | implicated in cellular migration and adhesion in the nervous system [33] |
| 1 | 50245–50798; | 553.125; | 73.14%; | LMBRD1 | participated in compound integral component of membrane [34] |
| 1 | 43257–450112; | 1754.52; | 72.17%; | SLC35F1 | play count role of control heat rate and cardiovascular disease [35] |
| 1 | 43257–45012; | 1754.52; | 72.17%; | DCBLD1 | participated in compound integral component of membrane [36] |
| 6 | 153543–153633; | 90.199; | 69.90%; | OMA1 | participated in compound integral component of membrane [37] |
| 8 | 86943–87935; 86188–86896; | 991.802; 708.867; | 68.28%; 66.67%; | CLGN | important modulator in spermatogenesis and infertility [38] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, H.; Zhang, S.; Zhang, K.; Peng, X.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Genome-Wide Patterns of Homozygosity and Relevant Characterizations on the Population Structure in Piétrain Pigs. Genes 2020, 11, 577. https://doi.org/10.3390/genes11050577
Zhan H, Zhang S, Zhang K, Peng X, Xie S, Li X, Zhao S, Ma Y. Genome-Wide Patterns of Homozygosity and Relevant Characterizations on the Population Structure in Piétrain Pigs. Genes. 2020; 11(5):577. https://doi.org/10.3390/genes11050577
Chicago/Turabian StyleZhan, Huiwen, Saixian Zhang, Kaili Zhang, Xia Peng, Shengsong Xie, Xinyun Li, Shuhong Zhao, and Yunlong Ma. 2020. "Genome-Wide Patterns of Homozygosity and Relevant Characterizations on the Population Structure in Piétrain Pigs" Genes 11, no. 5: 577. https://doi.org/10.3390/genes11050577
APA StyleZhan, H., Zhang, S., Zhang, K., Peng, X., Xie, S., Li, X., Zhao, S., & Ma, Y. (2020). Genome-Wide Patterns of Homozygosity and Relevant Characterizations on the Population Structure in Piétrain Pigs. Genes, 11(5), 577. https://doi.org/10.3390/genes11050577