rps3 as a Candidate Mitochondrial Gene for the Molecular Identification of Species from the Colletotrichum acutatum Species Complex

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Isolate and DNA Extraction
2.2. DNA Sequencing, Assembly, and Annotation of the Mitogenomes
2.3. Characteristic and Comparative Analysis of the Mitogenomes
2.4. Identification of Repetitive Elements
2.5. Phylogenetic Analysis
3. Results
3.1. Genome Size and Organization
3.2. Repeated Elements
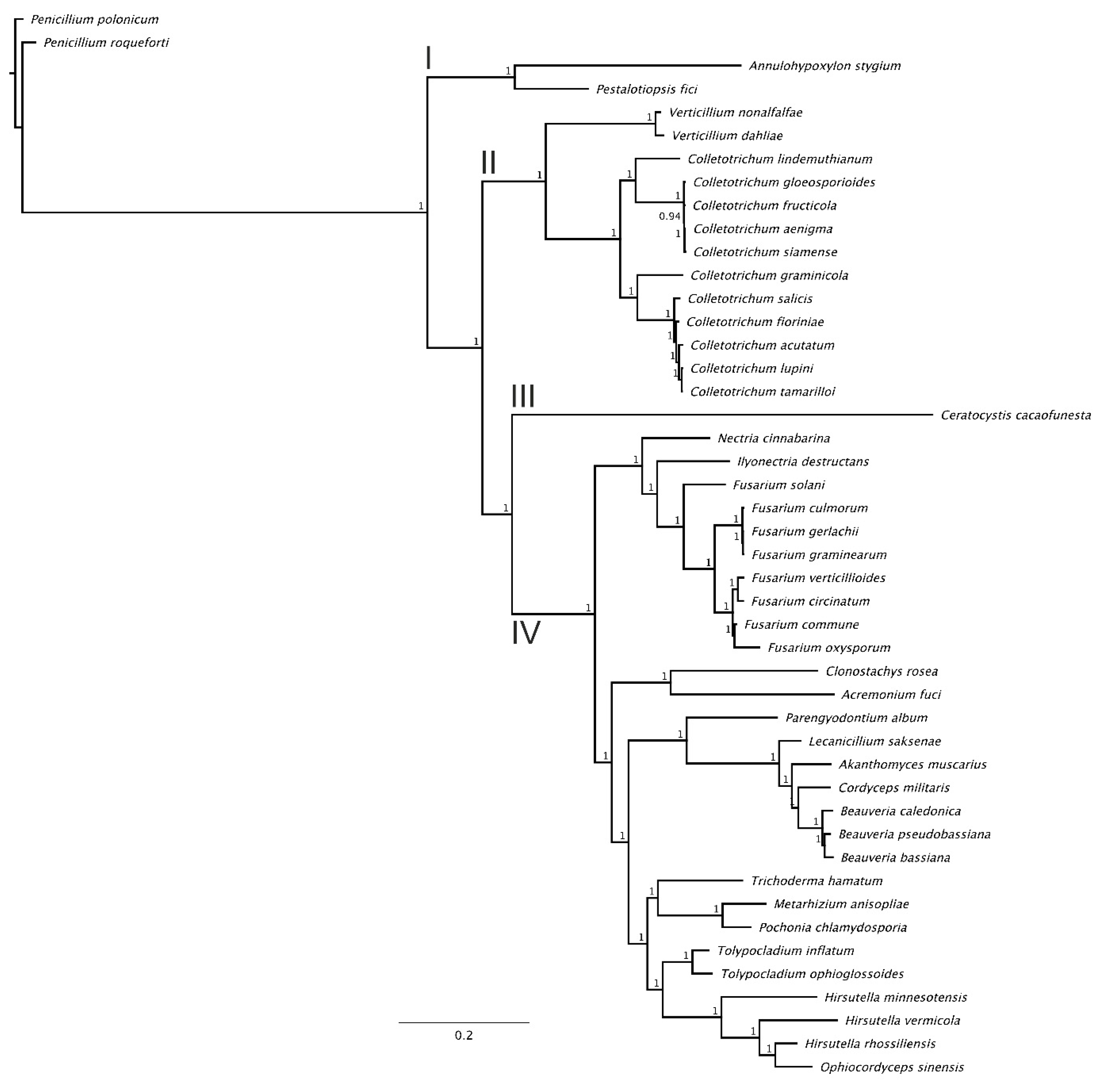
3.3. Phylogenomic Analysis
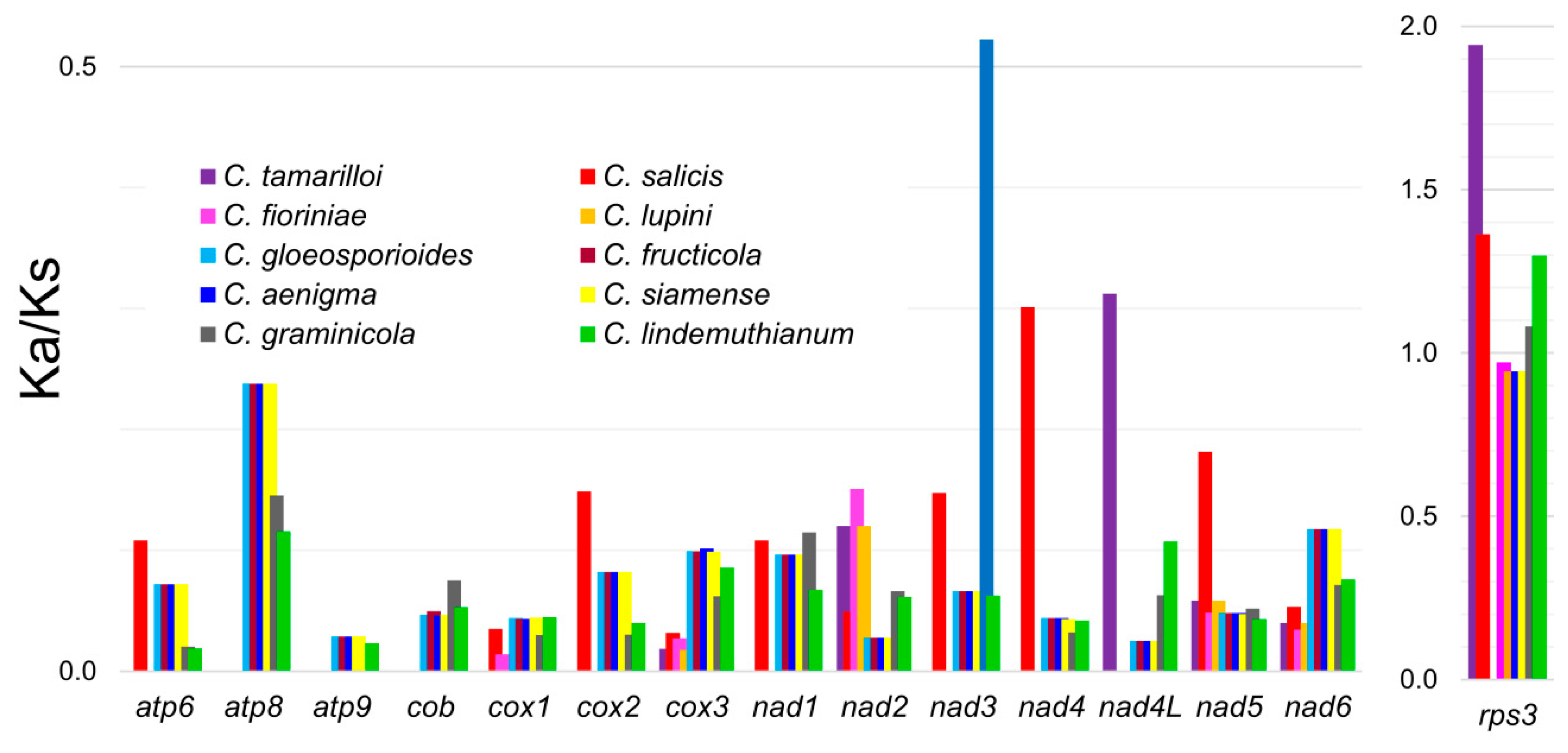
3.4. Molecular Evolution of rps3 in Colletotrichum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef]
- Sreenivasaprasad, S.; Talhinhas, P. Genotypic and phenotypic diversity in Colletotrichum acutatum, a cosmopolitan pathogen causing anthracnose on a wide range of hosts. Mol. Plant Pathol. 2005, 6, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, R.S.; Hyde, K.D.; Jeewon, R.; Li, X.H.; Liu, M.; Yan, J.Y. Mycosphere Essay 6: Why is it important to correctly name Colletotrichum species? Mycosphere 2016, 7, 1076–1092. [Google Scholar] [CrossRef]
- Farr, D.F.; Rossman, A.Y. Fungal Databases, U.S. National Fungus Collections, ARS, USDA. Available online: https://nt.ars-grin.gov/fungaldatabases (accessed on 20 September 2019).
- Cannon, P.F.; Damm, U.; Johnston, P.R.; Weir, B.S. Colletotrichum current status and future directions. Stud. Mycol. 2012, 73, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Damm, U.; Cannon, P.F.; Woudenberg, J.H.C.; Crous, P.W. The Colletotrichum acutatum species complex. Stud. Mycol. 2012, 73, 37–113. [Google Scholar] [CrossRef] [PubMed]
- Prusky, D.; Alkan, N.; Mengiste, T.; Fluhr, R. Quiescent and necrotrophic lifestyle choice during postharvest disease development. Annu. Rev. Phytopathol. 2013, 51, 155–176. [Google Scholar] [CrossRef]
- Prusky, D. Pathogen quiescence in postharvest diseases. Annu. Rev. Phytopathol. 1996, 34, 413–434. [Google Scholar] [CrossRef]
- Von Arx, J.A. Die Arten der Gattung Colletotrichum. Phytopathol. Z. 1957, 29, 413–468. [Google Scholar]
- Sutton, B.C. The genus Glomerella and its anamorph Colletotrichum. In Colletotrichum: Biology, Pathology, and Control; Bailey, J.A., Jeger, M.J., Eds.; CAB International: Wallingford, UK, 1992; pp. 1–26. [Google Scholar]
- Index Fungorum. Available online: https://indexfungorum.org/names/Names.asp (accessed on 19 November 2019).
- Vargas, W.A.; Martín, J.M.; Rech, G.E.; Rivera, L.P.; Benito, E.P.; Díaz-Mínguez, J.M.; Thon, M.R.; Sukno, S.A. Plant defense mechanisms are activated during biotrophic and necrotrophic development of Colletotricum graminicola in maize. Plant Physiol. 2012, 158, 1342–1358. [Google Scholar] [CrossRef]
- Talhinhas, P.; Baroncelli, R.; Le Floch, G. Anthracnose of lupins caused by Colletotrichum lupini: A recent disease and a successful worldwide pathogen. J. Plant Pathol. 2016, 98, 5–14. [Google Scholar]
- Pszczółkowska, A.; Okorski, A.; Paukszto, Ł.; Jastrzębski, J.P. First report of anthracnose disease caused by Colletotrichum fioriniae on blueberry in Western Poland. Plant Dis. 2016, 100, 2167. [Google Scholar] [CrossRef]
- Pszczółkowska, A.; Okorski, A.; Paukszto, Ł.; Jastrzębski, J.P.; Gorzkowska, A.; Chareńska, A. First report of Fagus sylvatica leaf spot infection by Colletotrichum fioriniae in forest nurseries in Northeastern Poland. Plant Dis. 2017, 101, 1822. [Google Scholar] [CrossRef]
- Pszczółkowska, A.; Okorski, A.; Kotecki, A.; Gas, M.; Kulik, T.; Reczek, A. Incidence of seed-borne fungi on Lupinus mutabilis depending on a plant morphotype, sowing date and plant density. J. Elem. 2016, 21, 501–512. [Google Scholar] [CrossRef][Green Version]
- Pszczółkowska, A.; Okorski, A.; Fordoński, G.; Prusiński, J.; Faligowska, A.; Borowska, M. Fungal colonization of seeds of three lupine species in different regions of Poland. Acta Agrobot. 2017, 70, 1714. [Google Scholar] [CrossRef]
- Okorski, A.; Pszczółkowska, A.; Sulima, P.; Paukszto, Ł.; Jastrzębski, J.P.; Przyborowski, J.; Makowczenko, K.G. First report of willow anthracnose caused by Colletotrichum salicis in Poland. Plant Dis. 2018, 102, 2036. [Google Scholar] [CrossRef]
- Baroncelli, R.; Amby, D.B.; Zapparata, A.; Sarrocco, S.; Vannacci, G.; Le Floch, G.; Harrison, R.J.; Holub, E.; Sukno, S.A.; Sreenivasaprasad, S.; et al. Gene family expansions and contractions are associated with host range in plant pathogens of the genus Colletotrichum. BMC Genom. 2016, 17, 555. [Google Scholar] [CrossRef]
- Baroncelli, R.; Talhinhas, P.; Pensec, F.; Sukno, S.A.; Le Floch, G.; Thon, M.R. The Colletotrichum acutatum species complex as a model system to study evolution and host specialization in plant pathogens. Front. Microbiol. 2017, 8, 2001. [Google Scholar] [CrossRef]
- Xu, J. Fungal DNA barcoding. Genome 2016, 59, 913–932. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- MFannot Tool. Available online: https://megasun.bch.umontreal.ca/cgi-bin/dev_mfa/mfannotInterface.pl (accessed on 6 May 2020).
- Dong, X.; Stothard, P.; Forsythe, I.J.; Wishart, D.S. PlasMapper: A webserver for drawing and auto-annotating plasmid maps. Nucleic Acids Res. 2004, 32, W660–W664. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Choi, K.Y.; Han, J.H.; Choi, I.Y.; Lee, Y.H.; Kim, K.S. The complete mitochondrial genome sequence of the ascomycete plant pathogen Colletotrichum acutatum. Mitochondrial DNA Part A 2016, 27, 4547–4548. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Tian, X.; Liu, W.; Wei, T.; Wang, W.; Dong, Q.; Wang, B.; Meng, Y.; Zhang, R.; Gleason, M.L.; et al. Comparative analysis of the mitochondrial genomes of Colletotrichum gloeosporioides sensu lato: Insights into the evolution of a fungal species complex interacting with diverse plants. BMC Genom. 2017, 18, 171. [Google Scholar] [CrossRef]
- Vaillancourt, L.; Ma, L.-J.; Thon, M.; O’Connell, R.; van Themaat, E.V.L.; Young, S.K.; Zeng, Q.; Gargeya, S.; Alvarado, L.; Berlin, A.; et al. Direct Submission; Broad Institute of MIT and Harvard, 7 Cambridge Center: Cambridge, MA, USA, 2015. [Google Scholar]
- Gutierrez, P.; Alzate, J.; Yepes, M.S.; Marin, M. Complete mitochondrial genome sequence of the common bean anthracnose pathogen Colletotrichum lindemuthianum. Mitochondrial DNA Part A 2016, 27, 136–137. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Del Barrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Mayer, C. Phobos 3.3.11. 2006–2010. Available online: http://www.rub.de/spezzoo/cm/cm_phobos.htm (accessed on 12 November 2019).
- Sablok, G.; Raju, G.V.P.; Mudunuri, S.B.; Prabha, R.; Singh, D.P.; Baev, V.; Yahubyan, G.; Ralph, P.J.; La Porta, N. ChloroMitoSSRDB 2.00: More genomes, more repeats, unifying SSRs search patterns and on-the-fly repeat detection. Database 2015, 2015, bav084. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- NCBI Website. Organelle Genome Resources. Available online: https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/ (accessed on 14 September 2019).
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Fourie, G.; van der Merwe, N.A.; Wingfield, B.D.; Bogale, M.; Tudzynski, B.; Wingfield, M.J.; Steenkamp, E.T. Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genom. 2013, 14, 605. [Google Scholar] [CrossRef]
- Salavirta, H.; Oksanen, I.; Kuuskeri, J.; Mäkelä, M.; Laine, P.; Paulin, L.; Lundell, T. Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among fungi and features signs of genome flexibility and recent recombination events. PLoS ONE 2014, 9, e97141. [Google Scholar] [CrossRef]
- Torriani, S.F.F.; Penselin, D.; Knogge, W.; Felder, M.; Taudien, S.; Platzer, M.; McDonald, B.A.; Brunner, P.C. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet. Biol. 2014, 62, 34–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Zhang, G.; Liu, X.; Wang, C.; Xu, J. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet. Biol. 2015, 77, 95–107. [Google Scholar] [CrossRef]
- Bullerwell, C.E.; Lang, B.F. Fungal evolution: The case of the vanishing mitochondrion. Curr. Opin. Microbiol. 2005, 8, 362–369. [Google Scholar] [CrossRef]
- Aguileta, G.; de Vienne, D.M.; Ross, O.N.; Hood, M.E.; Giraud, T.; Petit, E.; Gabaldón, T. High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 2014, 6, 451–465. [Google Scholar] [CrossRef]
- Lambowitz, A.M.; Belfort, M. Introns as mobile genetic elements. Annu. Rev. Biochem. 1993, 62, 587–622. [Google Scholar] [CrossRef]
- Saguez, C.; Lecellier, G.; Koll, F. Intronic GIY-YIG endonuclease gene in the mitochondrial genome of Podospora curvicolla: Evidence for mobility. Nucleic Acids Res. 2000, 28, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983. [Google Scholar]
- Makalowski, W.; Boguski, M.S. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2,820 orthologous rodent and human sequences. Proc. Natl. Acad. Sci. USA 1998, 95, 9407–9412. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Liu, C.; Shen, B.; Bai, M.; Ling, J.; Chen, G.; Mao, Z.; Cheng, X.; Xie, B. Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 2015, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, P.; Butow, R.A. The role of var1 in the assembly of yeast mitochondrial ribosomes. J. Biol. Chem. 1979, 254, 12662–12669. [Google Scholar] [PubMed]
- Terpstra, P.; Zanders, E.; Butow, R.A. The association of var1 with the 38S mitochondrial ribosomal subunit in yeast. J. Biol. Chem. 1979, 254, 2653–12661. [Google Scholar]
- Sethuraman, J.; Majer, A.; Iranpour, M.; Hausner, G. Molecular evolution of the mtDNA encoded rps3 gene among filamentous ascomycetes fungi with an emphasis on the ophiostomatoid fungi. J. Mol. Evol. 2009, 69, 372–385. [Google Scholar] [CrossRef]
- Wooll, G. Extraribosomal functions of ribosomal proteins. Trends Biochem. Sci. 1996, 21, 164–165. [Google Scholar] [CrossRef]
- Wai, A.; Shen, C.; Carta, A.; Dansen, A.; Crous, P.W.; Hausner, G. Intronencoded ribosomal proteins and N-acetyltransferases within the mitochondrial genomes of fungi: Here today, gone tomorrow? Mitochondrial DNA Part A 2019, 30, 573–584. [Google Scholar] [CrossRef]
- Korovesi, A.G.; Ntertilis, M.; Kouvelis, V.N. Mt-rps3 is an ancient gene which provides insight into the evolution of fungal mitochondrial genomes. Mol. Phylogenet. Evol. 2018, 127, 74–86. [Google Scholar] [CrossRef]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef]
- Kuramae, E.E.; Robert, V.; Snel, B.; Weiß, M.; Boekhout, T. Phylogenomics reveal a robust fungal tree of life. FEMS Yeast Res. 2006, 6, 6–1213. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D.S.; Binder, M.; Bischoff, J.F.; Blackwell, M.; Cannon, P.F.; Eriksson, O.E.; Huhndorf, S.; James, T.; Kirk, P.M.; Lucking, R.; et al. A higher-level phylogenetic classification of the fungi. Mycol. Res. 2007, 111, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Hu, L.; Shen, P.; Li, R.; Liu, D. SMRT Sequencing Revealed Mitogenome Characteristics and Mitogenome-Wide DNA Modification Pattern in Ophiocordyceps sinensis. Front. Microbiol. 2017, 8, 1422. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasaprasad, S.; Brown, A.E.; Mills, P.R. DNA sequence variation and interrelationship among Colletotrichum species causing strawberry anthracnose. Physiol. Mol. Plant Pathol. 1992, 41, 265–281. [Google Scholar] [CrossRef]
- Crouch, J.A.; Clarke, B.B.; Hillman, B.I. What is the value of ITS sequence data in Colletotrichum systematics and species diagnosis? A case study using the falcate-spored graminicolous Colletotrichum group. Mycologia 2009, 101, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Vieira, W.A.S.; Lima, W.G.; Nascimento, E.S.; Michereff, S.J.; Camara, M.P.S.; Doyle, V.P. The impact of phenotypic and molecular data on the inference of Colletotrichum diversity associated with Musa. Mycologia 2017, 109, 912–934. [Google Scholar] [CrossRef]
- Hyde, K.D.; Cai, L.; McKenzie, E.H.C.; Yang, Y.L.; Zhang, J.Z.; Prihastuti, H. Colletotrichum: A catalogue of confusion. Fungal Divers. 2009, 39, 1–17. [Google Scholar]
- Liu, F.; Wang, M.; Damm, U.; Crous, P.W.; Cai, L. Species boundaries in plant pathogenic fungi: A Colletotrichum case study. BMC Evol. Biol. 2016, 16, 81. [Google Scholar] [CrossRef]
- Vieira, W.A.S.; Bezerra, P.A.; da Silva, A.C.; Veloso, J.S.; Câmara, M.P.S.; Doyle, V.P. Optimal markers for the identification of Colletotrichum species. Mol. Phylogenet. Evol. 2020, 143, 106694. [Google Scholar] [CrossRef]
- Kane, N.C.; Cronk, Q. Botany without borders: Barcoding in focus. Mol. Ecol. 2008, 17, 5175–5517. [Google Scholar] [CrossRef]
- Du, M.; Schardl, C.L.; Nuckles, E.M.; Vaillancourt, L.J. Using mating-type gene sequences for improved phylogenetic resolution of Collectotrichum species complexes. Mycologia 2005, 97, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Hyde, K.D.; Taylor, P.W.J.; Weir, B.; Waller, J.; Abang, M.M.; Zhang, Z.J.; Yang, Y.L.; Phoulivong, S.; Liu, Z.Y.; et al. A polyphasic approach for studying Colletotrichum. Fungal Divers. 2009, 39, 183–204. [Google Scholar]
- Sharma, G.; Kumar, N.; Weir, B.S.; Hyde, K.D.; Shenoy, B.D. The ApMat marker can resolve Colletotrichum species: A case study with Mangifera indica. Fungal Divers. 2013, 61, 117–138. [Google Scholar] [CrossRef]
- Baroncelli, R.; Sanz-MartõÂn, J.M.; Rech, G.E.; Sukno, S.A.; Thon, M.R. Draft genome sequence of Colletotrichum sublineola, a destructive pathogen of cultivated sorghum. Genome Announc. 2014, 2, e00540-14. [Google Scholar] [CrossRef]
- Pszczółkowska, A.; Okorski, A.; Jastrzębski, J.P.; Paukszto, Ł.; Fordoński, G. The complete mitogenome of Colletotrichum lupini var. setosum. Mitochondrial DNA Part B 2016, 1, 41–42. [Google Scholar]
- Gan, P.; Narusaka, M.; Kumakura, N.; Tsushima, A.; Takano, Y.; Narusaka, Y. Genus-wide comparative genome analyses of Colletotrichum species reveal specific gene family losses and gains during adaptation to specific infection lifestyles. Genome Biol. Evol. 2016, 8, 1467–1481. [Google Scholar] [CrossRef]
- Gan, P.; Narusaka, M.; Tsushima, A.; Narusaka, Y.; Takano, Y.; Shirasu, K. Draft genome assembly of Colletotrichum chlorophyti, a pathogen of herbaceous plants. Genome Announc. 2017, 5, e01733-16. [Google Scholar] [CrossRef]
- Queiroz, C.B.; de Correia, H.L.N.; Menicucci, R.P.; Vidigal, P.M.P.; de Queiroz, M.V. Draft genome sequences of two isolates of Colletotrichum lindemuthianum, the causal agent of anthracnose in common beans. Genome Announc. 2017, 5, e00214–e00217. [Google Scholar] [CrossRef]
- Liang, X.; Wang, B.; Dong, Q.; Li, L.; Rollins, J.A.; Zhang, R.; Sun, G. Pathogenic adaptations of Colletotrichum fungi revealed by genome wide gene family evolutionary analyses. PLoS ONE 2018, 13, e0196303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Species | Complex | Accession number | Reference |
|---|---|---|---|
| C. acutatum | acutatum | NC_027280 | Kim et al. 2016 [27] |
| C. lupini | NC_029213 | This study | |
| C. fioriniae | NC_030052 | This study | |
| C. salicis | NC_035496 | This study | |
| C. tamarilloi | NC_029706 | This study | |
| C. gloeosporioides | gloeosporioides | KX885104 | Liang et al. 2017 [28] |
| C. aenigma | KX885105 | Liang et al. 2017 [28] | |
| C. fructicola | KX034082 | Liang et al. 2017 [28] | |
| C. siamense | KX885102 | Liang et al. 2017 [28] | |
| C. graminicola | graminicola | NW_007361658 | Vaillancourt et al. 2015 [29] |
| C. lindemuthianum | orbiculare | NC_023540 | Gutierrez et al. 2016 [30] |
| Species | Length [bp] | %GC | Start codon | Stop codon | PCGs | rRNAs | tRNAs | Introns | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | CDS | AUG | UUG | AUU | UAA | UAG | |||||||
| C. acutatum | 30,892 | 14,424 | 30.51 | 15 | 0 | 0 | 14 | 1 (nad6) | 15 | 2 | 28 | 1 | |
| C. lupini | 36,554 | 14,421 | 29.91 | 15 | 0 | 0 | 14 | 1 (nad6) | 15 | 2 | 29 | 1 | |
| C. fioriniae | 30,020 | 14,436 | 30.04 | 15 | 0 | 0 | 14 | 1 (nad6) | 15 | 2 | 29 | 1 | |
| C. salicis | 33,950 | 14,424 | 30.44 | 15 | 0 | 0 | 14 | 1 (nad6) | 15 | 2 | 29 | 2 | |
| C. tamarilloi | 30,824 | 14,421 | 30.50 | 15 | 0 | 0 | 14 | 1 (nad6) | 15 | 2 | 29 | 1 | |
| C. gloeosporioides | 55,169 | 14,721 | 34.55 | 14 | 1 (cox3) | 0 | 13 | 2 (cox3 and nad6) | 15 | 2 | 27 | 8 | |
| C. aenigma | 57,252 | 14,748 | 34.28 | 14 | 1 (cox3) | 0 | 13 | 2 (cox3 and nad6) | 15 | 2 | 27 | 9 | |
| C. fructicola | 56,051 | 14,475 | 34.04 | 14 | 1 (cox3) | 0 | 13 | 2 (cox3 and nad6) | 15 | 2 | 27 | 9 | |
| C. siamense | 54,645 | 14,745 | 34.30 | 14 | 1 (cox3) | 0 | 13 | 2 (cox3 and nad6) | 15 | 2 | 27 | 7 | |
| C. graminicola | 39,649 | 16,482 | 29.89 | 15 | 0 | 1 (atp6) | 15 | 1 (nad6) | 16 | 2 | 25 | 2 | |
| C. lindemuthianum | 36,957 | 15,012 | 30.88 | 15 | 0 | 1 | 14 | 2 (cox1 and nad6) | 16 | 2 | 27 | 2 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pszczółkowska, A.; Androsiuk, P.; Jastrzębski, J.P.; Paukszto, Ł.; Okorski, A. rps3 as a Candidate Mitochondrial Gene for the Molecular Identification of Species from the Colletotrichum acutatum Species Complex. Genes 2020, 11, 552. https://doi.org/10.3390/genes11050552
Pszczółkowska A, Androsiuk P, Jastrzębski JP, Paukszto Ł, Okorski A. rps3 as a Candidate Mitochondrial Gene for the Molecular Identification of Species from the Colletotrichum acutatum Species Complex. Genes. 2020; 11(5):552. https://doi.org/10.3390/genes11050552
Chicago/Turabian StylePszczółkowska, Agnieszka, Piotr Androsiuk, Jan Paweł Jastrzębski, Łukasz Paukszto, and Adam Okorski. 2020. "rps3 as a Candidate Mitochondrial Gene for the Molecular Identification of Species from the Colletotrichum acutatum Species Complex" Genes 11, no. 5: 552. https://doi.org/10.3390/genes11050552
APA StylePszczółkowska, A., Androsiuk, P., Jastrzębski, J. P., Paukszto, Ł., & Okorski, A. (2020). rps3 as a Candidate Mitochondrial Gene for the Molecular Identification of Species from the Colletotrichum acutatum Species Complex. Genes, 11(5), 552. https://doi.org/10.3390/genes11050552