Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk



 ,
,  ,
,  and
and 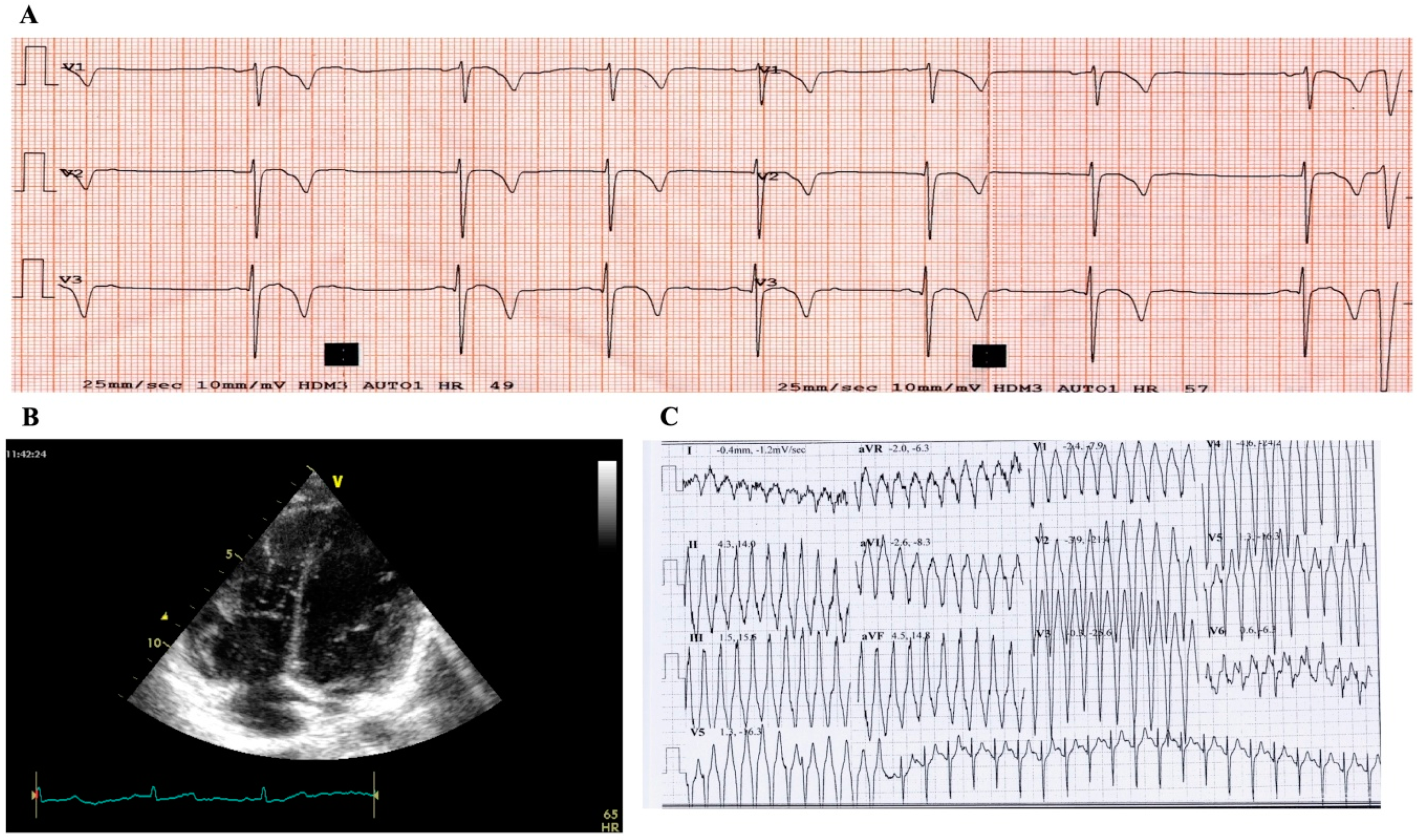{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Case Study Presentation and Clinical Description
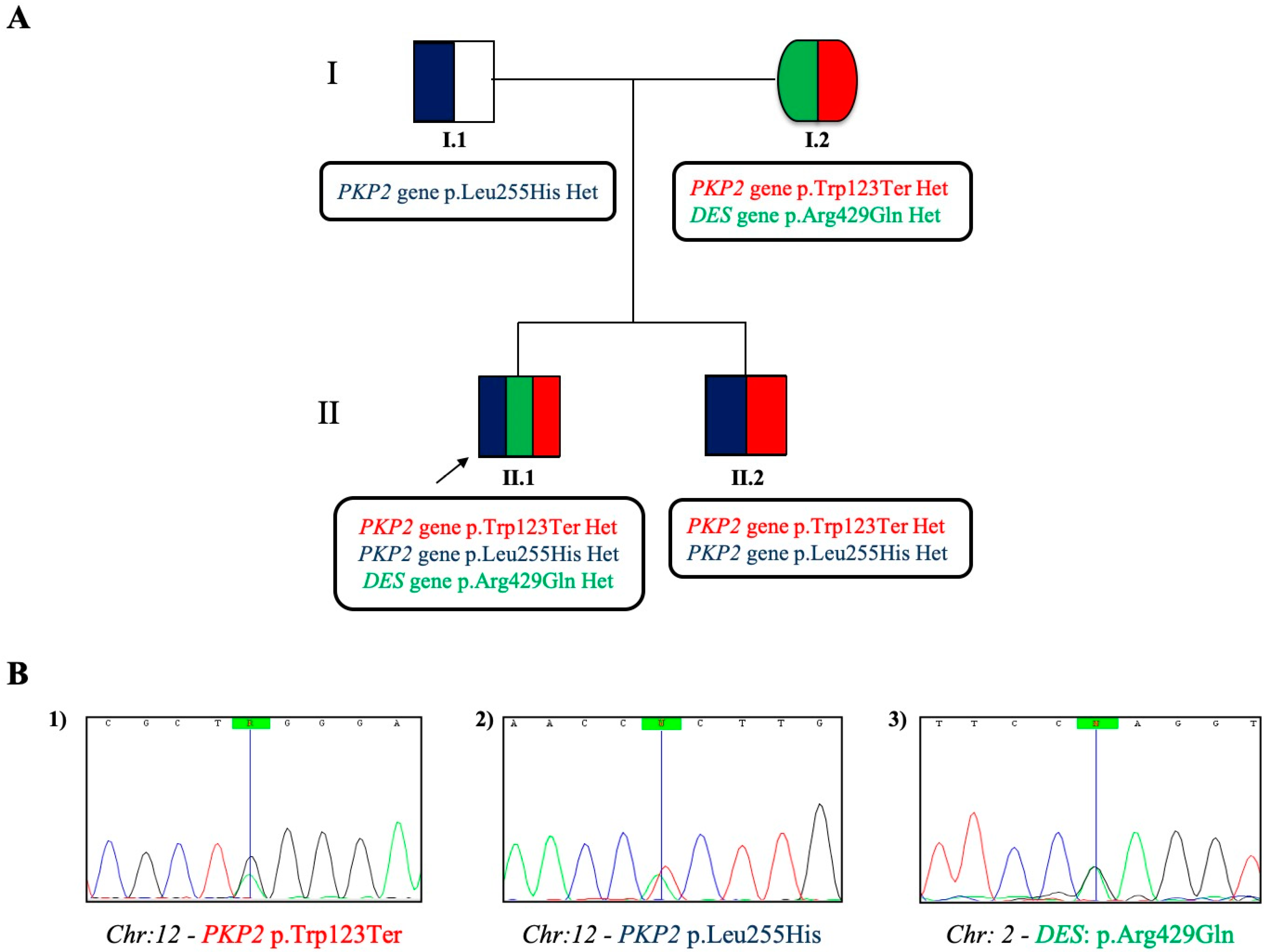
2.2. Case Study: Clinical Molecular Biology Investigation
3. Discussion
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corrado, D.; Schmied, C.; Basso, C.; Borjesson, M.; Schiavon, M.; Pelliccia, A.; Vanhees, L.; Thiene, G. Risk of sports: Do we need a pre-participation screening for competitive and leisure athletes? Eur. Heart J. 2011, 32, 934–944. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Pavei, A.; Michieli, P.; Schiavon, M.; Thiene, G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA 2006, 296, 1593–1601. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef] [PubMed]
- Lawley, C.M.; Skinner, J.R.; Turner, C. Syncope Due to Ventricular Arrhythmia Triggered by Electronic Gaming. N. Engl. J. Med. 2019, 381, 1180–1181. [Google Scholar] [CrossRef]
- Cirino, A.L.; Harris, S.; Lakdawala, N.K.; Michels, M.; Olivotto, I.; Day, S.M.; Abrams, D.J.; Charron, P.; Caleshu, C.; Semsarian, C.; et al. Role of genetic testing in inherited cardiovascular disease: A review. JAMA Cardiol. 2017, 2, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Biagini, E.; Olivotto, I.; Iascone, M.; Parodi, M.I.; Girolami, F.; Frisso, G.; Autore, C.; Limongelli, G.; Cecconi, M.; Maron, B.J.; et al. Significance of sarcomere gene mutations analysis in the end-stage phase of hypertrophic cardiomyopathy. Am. J. Cardiol. 2014, 114, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Carsana, A.; Frisso, G.; Tremolaterra, M.R.; Ricci, E.; De Rasmo, D.; Salvatore, F. A larger spectrum of intragenic short tandem repeats improves linkage analysis and localization of intragenic recombination detection in the dystrophin gene. An analysis of 93 families from Southern Italy. J. Mol. Diagn. 2007, 9, 64–69. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frisso, G.; Limongelli, G.; Pacileo, G.; Del Giudice, A.; Forgione, L.; Calabrò, P.; Iacomino, M.; Detta, N.; Di Fonzo, L.M.; Maddaloni, V.; et al. A child cohort study from southern Italy enlarges the genetic spectrum of hypertrophic cardiomyopathy. Clin. Genet. 2009, 76, 91–101. [Google Scholar] [CrossRef]
- Aneq, M.Å.; Fluur, C.; Rehnberg, M.; Söderkvist, P.; Engvall, J.; Nylander, E.; Gunnarsson, C. Novel plakophilin2 mutation: Three-generation family with arrhythmogenic right ventricular cardiomyopathy. Scand. Cardiovasc. J. 2012, 46, 72–75. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Frisso, G.; Precone, V.; Boccia, A.; Fienga, A.; Pacileo, G.; Limongelli, G.; Paolella, G.; Calabrò, R.; Salvatore, F. DNA sequence capture and next-generation sequencing for the molecular diagnosis of genetic cardiomyopathies. J. Mol. Diagn. 2014, 16, 32–44. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Esposito, M.V.; Nunziato, M.; De Simone, A.; Buono, P.; Salvatore, F.; Frisso, G. Molecular diagnosis of Brugada syndrome via next-generation sequencing of a multigene panel in a young athlete Diagnosi molecolare di sindrome di Brugada in un. Med. Dello Sport 2018, 71, 27–34. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Rizzoli, G.; Schiavon, M.; Thiene, G. Does Sports Activity Enhance the Risk of Sudden Death in Adolescents and Young Adults? J. Am. Coll. Cardiol. 2003, 42, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G. Sudden death in young athletes. In Arrhythmogenic RV Cardiomyopathy/Dysplasia: Recent Advances; Marcus, F.I., Nava, A., Thiene, G., Eds.; Springer: New York, NY, USA, 2007; ISBN 9788847004894. [Google Scholar]
- Ruwald, A.C.; Marcus, F.; Estes, N.A.M.; Link, M.; McNitt, S.; Polonsky, B.; Calkins, H.; Towbin, J.A.; Moss, A.J.; Zareba, W. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: Results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopath. Eur. Heart J. 2015, 36, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Saberniak, J.; Hasselberg, N.E.; Borgquist, R.; Platonov, P.G.; Sarvari, S.I.; Smith, H.J.; Ribe, M.; Holst, A.G.; Edvardsen, T.; Haugaa, K.H. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur. J. Heart Fail. 2014, 16, 1337–1344. [Google Scholar] [CrossRef]
- Detta, N.; Frisso, G.; Limongelli, G.; Marzullo, M.; Calabrò, R.; Salvatore, F. Genetic analysis in a family affected by sick sinus syndrome may reduce the sudden death risk in a young aspiring competitive athlete. Int. J. Cardiol. 2014, 170, e63–e65. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Redi, A.; Lemme, E.; Pelliccia, A.; Salvatore, F.; Frisso, G. Impact of molecular diagnostics in an asymptomatic amateur athlete found to be affected by hypertrophic cardiomyopathy. Med. Dello Sport 2018, 71, 405–412. [Google Scholar] [CrossRef]
- Walsh, R.; Cook, S.A. Issues and challenges in diagnostic sequencing for inherited cardiac conditions. Clin. Chem. 2017, 63, 116–128. [Google Scholar] [CrossRef]
- Garcia, J.; Tahiliani, J.; Johnson, N.M.; Aguilar, S.; Beltran, D.; Daly, A.; Decker, E.; Haverfield, E.; Herrera, B.; Murillo, L.; et al. Clinical Genetic Testing for the Cardiomyopathies and Arrhythmias: A Systematic Framework for Establishing Clinical Validity and Addressing Genotypic and Phenotypic Heterogeneity. Front. Cardiovasc. Med. 2016, 3. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. International Experts. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm 2019, 16, e373–e407. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Varley, I. Exploring the Regulation of Genetic Testing in Sport. Entertain. Sports Law J. 2019, 17, 5. [Google Scholar] [CrossRef]
- Cater, C.; MacDonald, M.; Lithwick, D.; Sidhu, K.; Isserow, S.; McKinney, J. Perspectives on pre-participation cardiovascular screening in young competitive athletes: U SPORTS. Phys. Sportsmed. 2018, 46, 509–514. [Google Scholar] [CrossRef]
- Chatard, J.C.; Mujika, I.; Goiriena, J.J.; Carré, F. Screening young athletes for prevention of sudden cardiac death: Practical recommendations for sports physicians. Scand. J. Med. Sci. Sport 2016, 26, 362–374. [Google Scholar] [CrossRef]
- Van Brabandt, H.; Desomer, A.; Gerkens, S.; Neyt, M. Harms and benefits of screening young people to prevent sudden cardiac death. BMJ 2016, 353. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Peters, S. Advances in the diagnostic management of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int. J. Cardiol. 2006, 113, 4–11. [Google Scholar] [CrossRef]
- Thiene, G.; Basso, C. Arrhythmogenic right ventricular cardiomyopathy: An update. Cardiovasc. Pathol. 2001, 10, 109–117. [Google Scholar] [CrossRef]
- Nejat, M.; Saedi, S.; Soveizi, M.; Rabbani, B.; Najafi, N.; Maleki, M. A novel PKP2 mutation and intrafamilial phenotypic variability in ARVC/D. Med. J. Islamic Repub. Iran 2018, 32, 5. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and Digenic Heterozygosity Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wang, J.; Yao, Y.; Wang, Y.; Fan, X.; Sun, K.; He, D.S.; Marcus, F.I.; Zhang, S.; Hui, R.; et al. Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; Te Riele, A.S.J.M.; Van Den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.P.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Quarta, G.; Muir, A.; Pantazis, A.; Syrris, P.; Gehmlich, K.; Garcia-Pavia, P.; Ward, D.; Sen-Chowdhry, S.; Elliott, P.M.; McKenna, W.J. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: Impact of genetics and revised task force criteria. Circulation 2011, 123, 2701–2709. [Google Scholar] [CrossRef]
- Esposito, M.V.; Minopoli, G.; Esposito, L.; D’Argenio, V.; Di Maggio, F.; Sasso, E.; D’Aiuto, M.; Zambrano, N.; Salvatore, F. A Functional Analysis of the Unclassified Pro2767Ser BRCA2 Variant Reveals Its Potential Pathogenicity that Acts by Hampering DNA Binding and Homology-Mediated DNA Repair. Cancers 2019, 11, 1454. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Di Maggio, F.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes 2020, 11, 524. https://doi.org/10.3390/genes11050524
Limongelli G, Nunziato M, Mazzaccara C, Intrieri M, D’Argenio V, Esposito MV, Monda E, Di Maggio F, Frisso G, Salvatore F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes. 2020; 11(5):524. https://doi.org/10.3390/genes11050524
Chicago/Turabian StyleLimongelli, Giuseppe, Marcella Nunziato, Cristina Mazzaccara, Mariano Intrieri, Valeria D’Argenio, Maria Valeria Esposito, Emanuele Monda, Federica Di Maggio, Giulia Frisso, and Francesco Salvatore. 2020. "Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk" Genes 11, no. 5: 524. https://doi.org/10.3390/genes11050524
APA StyleLimongelli, G., Nunziato, M., Mazzaccara, C., Intrieri, M., D’Argenio, V., Esposito, M. V., Monda, E., Di Maggio, F., Frisso, G., & Salvatore, F. (2020). Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes, 11(5), 524. https://doi.org/10.3390/genes11050524