Application of CRISPR Tools for Variant Interpretation and Disease Modeling in Inherited Retinal Dystrophies

 , and
, and
Abstract
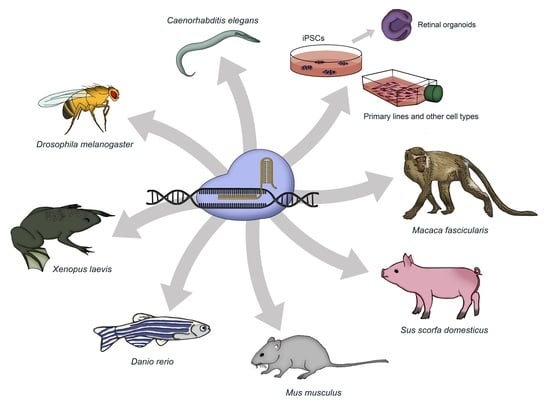
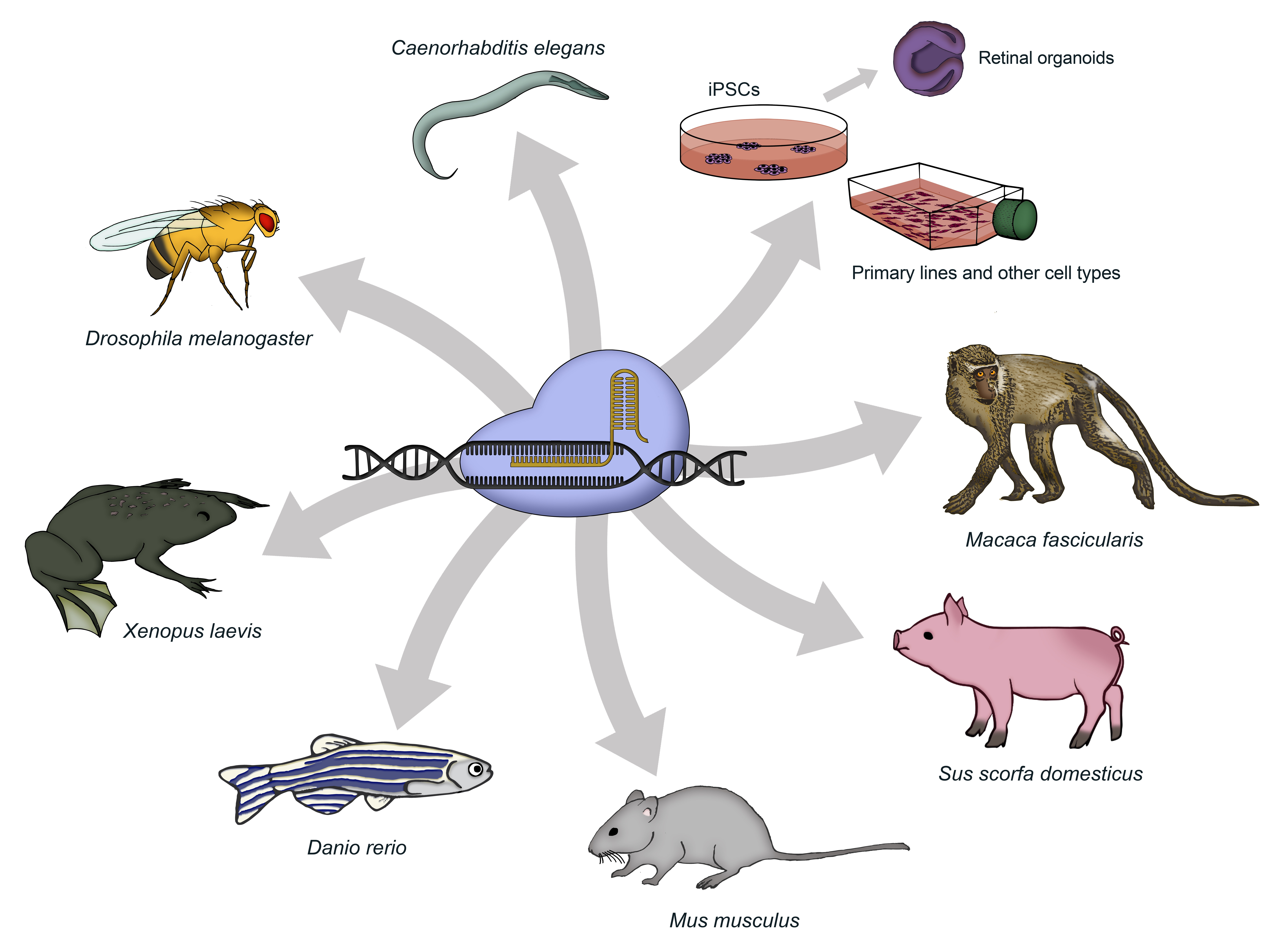
1. Introduction
2. The CRISPR Toolkit—Initial Steps
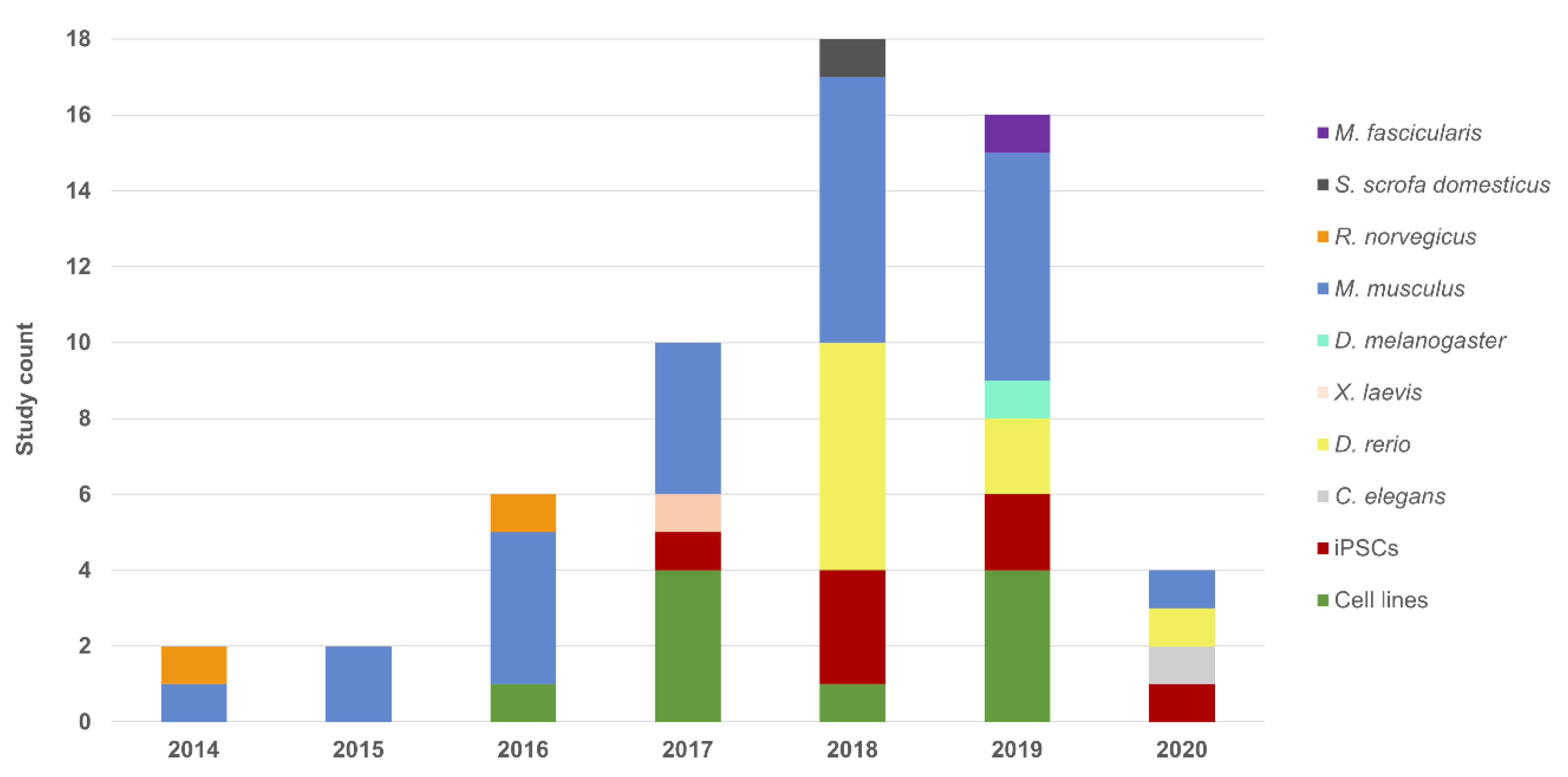
3. Cellular Models
3.1. Cell Lines
3.2. Induced Pluripotent Stem Cells
3.3. Retinal Organoids
4. Animal Models
4.1. Caenorhabditis elegans
4.2. Drosophila melanogaster
4.3. Xenopus
4.4. Zebrafish
4.5. Rodents
4.6. Pig
4.7. Macaque
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.E.; Day, M.A. Advantages and disadvantages of molecular testing in ophthalmology. Expert Rev. Ophthalmol. 2011, 6, 221–245. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Roepman, R.; Cremers, F.P.M.; den Hollander, A.I.; Mans, D.A. Non-syndromic retinal ciliopathies: Translating gene discovery into therapy. Hum. Mol. Genet. 2012, 21, R111–R124. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, O. Myron Yanoff and Jay S. Duker: Ophthalmology, Fifth Edition. Graefes Arch. Clin. Exp. Ophthalmol. 2020, 258, 459. [Google Scholar] [CrossRef]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef]
- Dreyer, B.; Tranebjaerg, L.; Brox, V.; Rosenberg, T.; Möller, C.; Beneyto, M.; Weston, M.D.; Kimberling, W.J.; Cremers, C.W.; Liu, X.Z.; et al. A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am. J. Hum. Genet. 2001, 69, 228–234. [Google Scholar]
- Seyedahmadi, B.J.; Rivolta, C.; Keene, J.A.; Berson, E.L.; Dryja, T.P. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp. Eye Res. 2004, 79, 167–173. [Google Scholar] [CrossRef]
- Rivera, A.; White, K.; Stöhr, H.; Steiner, K.; Hemmrich, N.; Grimm, T.; Jurklies, B.; Lorenz, B.; Scholl, H.P.; Apfelstedt-Sylla, E.; et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 2000, 67, 800–813. [Google Scholar] [CrossRef]
- Ziviello, C.; Simonelli, F.; Testa, F.; Anastasi, M.; Marzoli, S.B.; Falsini, B.; Ghiglione, D.; Macaluso, C.; Manitto, M.P.; Garrè, C.; et al. Molecular genetics of autosomal dominant retinitis pigmentosa (ADRP): A comprehensive study of 43 Italian families. J. Med. Genet. 2005, 42, e47. [Google Scholar] [CrossRef]
- Audo, I.; Manes, G.; Mohand-Saïd, S.; Friedrich, A.; Lancelot, M.-E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Zanlonghi, X.; Hamel, C.P.; et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3687–3700. [Google Scholar] [CrossRef]
- Valkenburg, D.; Van Cauwenbergh, C.; Lorenz, B.; van Genderen, M.M.; Bertelsen, M.; Pott, J.W.; Coppieters, F.; De Zaeytijd, J.; Thiadens, A.A.; Klaver, C.C.; et al. Clinical Characterization of 66 Patients With Congenital Retinal Disease Due to the Deep-Intronic c.2991+1655A>G Mutation in CEP290. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4384–4391. [Google Scholar] [CrossRef]
- Bernardis, I.; Chiesi, L.; Tenedini, E.; Artuso, L.; Percesepe, A.; Artusi, V.; Simone, M.L.; Manfredini, R.; Camparini, M.; Rinaldi, C.; et al. Unravelling the Complexity of Inherited Retinal Dystrophies Molecular Testing: Added Value of Targeted Next-Generation Sequencing. Biomed. Res. Int. 2016, 2016, 6341870. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- De Koning, T.J.; Jongbloed, J.D.H.; Sikkema-Raddatz, B.; Sinke, R.J. Targeted next-generation sequencing panels for monogenetic disorders in clinical diagnostics: The opportunities and challenges. Expert Rev. Mol. Diagn. 2015, 15, 61–70. [Google Scholar] [CrossRef]
- Lee, H.; Martinez-Agosto, J.A.; Rexach, J.; Fogel, B.L. Next generation sequencing in clinical diagnosis. Lancet Neurol. 2019, 18, 426. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Legut, M.; Daniloski, Z.; Xue, X.; McKenzie, D.; Guo, X.; Wessels, H.-H.; Sanjana, N.E. High-Throughput Screens of PAM-Flexible Cas9 Variants for Gene Knockout and Transcriptional Modulation. Cell Rep. 2020, 30, 2859–2868. [Google Scholar] [CrossRef]
- Gleditzsch, D.; Pausch, P.; Müller-Esparza, H.; Özcan, A.; Guo, X.; Bange, G.; Randau, L. PAM identification by CRISPR-Cas effector complexes: Diversified mechanisms and structures. RNA Biol. 2018, 16, 504–517. [Google Scholar] [CrossRef]
- Periwal, V. A comprehensive overview of computational resources to aid in precision genome editing with engineered nucleases. Brief. Bioinform. 2017, 18, 698–711. [Google Scholar] [CrossRef]
- Torres-Perez, R.; Garcia-Martin, J.A.; Montoliu, L.; Oliveros, J.C.; Pazos, F. WeReview: CRISPR Tools-Live Repository of Computational Tools for Assisting CRISPR/Cas Experiments. Bioengineering 2019, 6, 63. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-Y.; Sung, J.J.; Choi, S.-H.; Lee, D.R.; Park, I.-H.; Kim, D.-W. Modeling and correction of structural variations in patient-derived iPSCs using CRISPR/Cas9. Nat. Protoc. 2016, 11, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Brunet, E.; Jasin, M. Induction of Chromosomal Translocations with CRISPR-Cas9 and Other Nucleases: Understanding the Repair Mechanisms That Give Rise to Translocations. Adv. Exp. Med. Biol. 2018, 1044, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef]
- Horii, T.; Hatada, I. Production of genome-edited pluripotent stem cells and mice by CRISPR/Cas. Endocr. J. 2016, 63, 213–219. [Google Scholar] [CrossRef]
- Kurata, M.; Wolf, N.K.; Lahr, W.S.; Weg, M.T.; Kluesner, M.G.; Lee, S.; Hui, K.; Shiraiwa, M.; Webber, B.R.; Moriarity, B.S. Highly multiplexed genome engineering using CRISPR/Cas9 gRNA arrays. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.-H.; Suh, Y. In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Latella, M.C.; Salvo, M.T.D.; Cocchiarella, F.; Benati, D.; Grisendi, G.; Comitato, A.; Marigo, V.; Recchia, A. In vivo Editing of the Human Mutant Rhodopsin Gene by Electroporation of Plasmid-based CRISPR/Cas9 in the Mouse Retina. Mol. Ther. Nucleic Acids 2016, 5. [Google Scholar] [CrossRef]
- Ruan, G.-X.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.H.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. 2017, 25, 331–341. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-García, G.; González-Romero, E.; Jaijo, T.; Sequedo, M.D.; Ayuso, C.; Vázquez-Manrique, R.P.; Millán, J.M.; Aller, E. USH2A Gene Editing Using the CRISPR System. Mol. Ther. Nucleic Acids 2017, 8, 529–541. [Google Scholar] [CrossRef]
- Lv, J.-N.; Zhou, G.-H.; Chen, X.; Chen, H.; Wu, K.-C.; Xiang, L.; Lei, X.-L.; Zhang, X.; Wu, R.-H.; Jin, Z.-B. Targeted RP9 ablation and mutagenesis in mouse photoreceptor cells by CRISPR-Cas9. Sci. Rep. 2017, 7, 43062. [Google Scholar] [CrossRef]
- Lyraki, R.; Lokaj, M.; Soares, D.C.; Little, A.; Vermeren, M.; Marsh, J.A.; Wittinghofer, A.; Hurd, T. Characterization of a novel RP2-OSTF1 interaction and its implication for actin remodelling. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Zhang, Q.; Giacalone, J.C.; Searby, C.; Stone, E.M.; Tucker, B.A.; Sheffield, V.C. Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc. Natl. Acad. Sci. USA 2019, 116, 1353–1360. [Google Scholar] [CrossRef]
- Vagni, P.; Perlini, L.E.; Chenais, N.A.L.; Marchetti, T.; Parrini, M.; Contestabile, A.; Cancedda, L.; Ghezzi, D. Gene Editing Preserves Visual Functions in a Mouse Model of Retinal Degeneration. Front. Neurosci. 2019, 13, 1–18. [Google Scholar] [CrossRef]
- Jo, D.H.; Song, D.W.; Cho, C.S.; Kim, U.G.; Lee, K.J.; Lee, K.; Park, S.W.; Kim, D.; Kim, J.H.; Kim, J.S.; et al. CRISPR-Cas9–mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci. Adv. 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Yang, X.; Bayat, V.; DiDonato, N.; Zhao, Y.; Zarnegar, B.; Siprashvili, Z.; Lopez-Pajares, V.; Sun, T.; Tao, S.; Li, C.; et al. Genetic and genomic studies of pathogenic EXOSC2 mutations in the newly described disease SHRF implicate the autophagy pathway in disease pathogenesis. Hum. Mol. Genet. 2019, 29, 541–553. [Google Scholar] [CrossRef]
- Burnight, E.R.; Gupta, M.; Wiley, L.A.; Anfinson, K.R.; Tran, A.; Triboulet, R.; Hoffmann, J.M.; Klaahsen, D.L.; Andorf, J.L.; Jiao, C.; et al. Using CRISPR-Cas9 to Generate Gene-Corrected Autologous iPSCs for the Treatment of Inherited Retinal Degeneration. Mol. Ther. 2017, 25, 1999–2013. [Google Scholar] [CrossRef] [PubMed]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Brydon, E.M.; Bronstein, R.; Buskin, A.; Lako, M.; Pierce, E.A.; Fernandez-Godino, R. AAV-Mediated Gene Augmentation Therapy Restores Critical Functions in Mutant PRPF31+/− iPSC-Derived RPE Cells. Mol. Ther. Methods Clin. Dev. 2019, 15, 392–402. [Google Scholar] [CrossRef]
- Foltz, L.P.; Howden, S.E.; Thomson, J.A.; Clegg, D.O. Functional Assessment of Patient-Derived Retinal Pigment Epithelial Cells Edited by CRISPR/Cas9. Int. J. Mol. Sci. 2018, 19, 4127. [Google Scholar] [CrossRef]
- Deng, W.-L.; Gao, M.-L.; Lei, X.-L.; Lv, J.-N.; Zhao, H.; He, K.-W.; Xia, X.-X.; Li, L.-Y.; Chen, Y.-C.; Li, Y.-P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Bohrer, L.R.; Wiley, L.A.; Burnight, E.R.; Cooke, J.A.; Giacalone, J.C.; Anfinson, K.R.; Andorf, J.L.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9. Genes 2019, 10, 278. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Erkilic, N.; Baux, D.; Mamaeva, D.; Hamel, C.P.; Meunier, I.; Roux, A.-F.; Kalatzis, V. Genome Editing in Patient iPSCs Corrects the Most Prevalent USH2A Mutations and Reveals Intriguing Mutant mRNA Expression Profiles. Mol. Ther. Methods Clin. Dev. 2020, 17, 156–173. [Google Scholar] [CrossRef]
- Yang, T.-C.; Chang, C.-Y.; Yarmishyn, A.A.; Mao, Y.-S.; Yang, Y.-P.; Wang, M.-L.; Hsu, C.-C.; Yang, H.-Y.; Hwang, D.-K.; Chen, S.-J.; et al. Carboxylated nanodiamond-mediated CRISPR-Cas9 delivery of human retinoschisis mutation into human iPSCs and mouse retina. Acta Biomater. 2020, 101, 484–494. [Google Scholar] [CrossRef]
- Kukhtar, D.; Rubio-Peña, K.; Serrat, X.; Cerón, J. Mimicking of splicing-related retinitis pigmentosa mutations in C. elegans allow drug screens and identification of disease modifiers. Hum. Mol. Genet. 2020, 29, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Messchaert, M.; Dona, M.; Broekman, S.; Peters, T.A.; Corral-Serrano, J.C.; Slijkerman, R.W.N.; van Wijk, E.; Collin, R.W.J. Eyes shut homolog is important for the maintenance of photoreceptor morphology and visual function in zebrafish. PLoS ONE 2018, 13, e0200789. [Google Scholar] [CrossRef]
- Corral-Serrano, J.C.; Messchaert, M.; Dona, M.; Peters, T.A.; Kamminga, L.M.; van Wijk, E.; Collin, R.W.J. C2orf71a/pcare1 is important for photoreceptor outer segment morphogenesis and visual function in zebrafish. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Unal Eroglu, A.; Mulligan, T.S.; Zhang, L.; White, D.T.; Sengupta, S.; Nie, C.; Lu, N.Y.; Qian, J.; Xu, L.; Pei, W.; et al. Multiplexed CRISPR/Cas9 Targeting of Genes Implicated in Retinal Regeneration and Degeneration. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Minegishi, Y.; Nakaya, N.; Tomarev, S.I. Mutation in the Zebrafish cct2 Gene Leads to Abnormalities of Cell Cycle and Cell Death in the Retina: A Model of CCT2-Related Leber Congenital Amaurosis. Investig. Ophthalmol. Vis. Sci. 2018, 59, 995–1004. [Google Scholar] [CrossRef]
- Dona, M.; Slijkerman, R.; Lerner, K.; Broekman, S.; Wegner, J.; Howat, T.; Peters, T.; Hetterschijt, L.; Boon, N.; de Vrieze, E.; et al. Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. Eye Res. 2018, 173, 148–159. [Google Scholar] [CrossRef]
- Slijkerman, R.; Goloborodko, A.; Broekman, S.; de Vrieze, E.; Hetterschijt, L.; Peters, T.; Gerits, M.; Kremer, H.; van Wijk, E. Poor Splice-Site Recognition in a Humanized Zebrafish Knockin Model for the Recurrent Deep-Intronic c.7595-2144A>G Mutation in USH2A. Zebrafish 2018, 15, 597–609. [Google Scholar] [CrossRef]
- Xie, S.; Han, S.; Qu, Z.; Liu, F.; Li, J.; Yu, S.; Reilly, J.; Tu, J.; Liu, X.; Lu, Z.; et al. Knockout of Nr2e3 prevents rod photoreceptor differentiation and leads to selective L-/M-cone photoreceptor degeneration in zebrafish. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1273–1283. [Google Scholar] [CrossRef]
- Schlegel, D.K.; Glasauer, S.M.K.; Mateos, J.M.; Barmettler, G.; Ziegler, U.; Neuhauss, S.C.F. A New Zebrafish Model for CACNA2D4-Dysfunction. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5124–5135. [Google Scholar] [CrossRef]
- Karjosukarso, D.W.; Ali, Z.; Peters, T.A.; Zhang, J.Q.C.; Hoogendoorn, A.D.M.; Garanto, A.; van Wijk, E.; Jensen, L.D.; Collin, R.W.J. Modeling ZNF408-Associated FEVR in Zebrafish Results in Abnormal Retinal Vasculature. Investig. Ophthalmol. Vis. Sci. 2020, 61, 39. [Google Scholar] [CrossRef]
- Wang, S.; Sengel, C.; Emerson, M.M.; Cepko, C.L. A gene regulatory network controls the binary fate decision of rod and bipolar cells in the vertebrate retina. Dev. Cell 2014, 30, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, S.; Chen, H.Y.; Isgrig, K.; Yu, W.; Hiriyanna, S.; Levron, R.; Li, T.; Colosi, P.; Chien, W.; Swaroop, A.; et al. A CEP290 C-terminal Domain Complements the Mutant CEP290 of Rd16 Mice in Trans and Rescues Retinal Degeneration. Cell Rep. 2018, 25, 611–623. [Google Scholar] [CrossRef]
- Li, Y.; Furhang, R.; Ray, A.; Duncan, T.; Soucy, J.; Mahdi, R.; Chaitankar, V.; Gieser, L.; Poliakov, E.; Qian, H.; et al. Aberrant RNA splicing is the major pathogenic effect in a knock-in mouse model of the dominantly inherited c.1430A>G human RPE65 mutation. Hum. Mutat. 2019, 40, 426–443. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chen, Y.; Li, Y.; Chen, R.; Mardon, G. CRISPR-engineered mosaicism rapidly reveals that loss of Kcnj13 function in mice mimics human disease phenotypes. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Seruggia, D.; Fernández, A.; Cantero, M.; Pelczar, P.; Montoliu, L. Functional validation of mouse tyrosinase non-coding regulatory DNA elements by CRISPR-Cas9-mediated mutagenesis. Nucleic Acids Res. 2015, 43, 4855–4867. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.C.; Chrysostomou, V.; Li, F.; Lim, J.K.H.; Wang, J.-H.; Powell, J.E.; Tu, L.; Daniszewski, M.; Lo, C.; Wong, R.C.; et al. AAV-Mediated CRISPR/Cas Gene Editing of Retinal Cells In Vivo. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3470–3476. [Google Scholar] [CrossRef]
- Wu, W.-H.; Tsai, Y.-T.; Justus, S.; Lee, T.-T.; Zhang, L.; Lin, C.-S.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol. Ther. 2016, 24, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Arno, G.; Agrawal, S.A.; Eblimit, A.; Bellingham, J.; Xu, M.; Wang, F.; Chakarova, C.; Parfitt, D.A.; Lane, A.; Burgoyne, T.; et al. Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2016, 99, 1305–1315. [Google Scholar] [CrossRef]
- Agrawal, S.A.; Burgoyne, T.; Eblimit, A.; Bellingham, J.; Parfitt, D.A.; Lane, A.; Nichols, R.; Asomugha, C.; Hayes, M.J.; Munro, P.M.; et al. REEP6 deficiency leads to retinal degeneration through disruption of ER homeostasis and protein trafficking. Hum. Mol. Genet. 2017, 26, 2667–2677. [Google Scholar] [CrossRef]
- Moye, A.R.; Singh, R.; Kimler, V.A.; Dilan, T.L.; Munezero, D.; Saravanan, T.; Goldberg, A.F.X.; Ramamurthy, V. ARL2BP, a protein linked to retinitis pigmentosa, is needed for normal photoreceptor cilia doublets and outer segment structure. Mol. Biol. Cell 2018, 29, 1590–1598. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, Z.; Zhao, P.; Huang, L.; Xu, M.; Yang, Y.; Chen, X.; Lu, F.; Zhang, X.; Wang, H.; et al. Whole-exome sequencing revealed HKDC1 as a candidate gene associated with autosomal-recessive retinitis pigmentosa. Hum. Mol. Genet. 2018, 27, 4157–4168. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, S.G.; Luoni, M.; Castoldi, V.; Massimino, L.; Cabassi, T.; Angeloni, D.; Demontis, G.C.; Leocani, L.; Andreazzoli, M.; Broccoli, V. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 2018, 27, 761–779. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-T.; Wu, W.-H.; Lee, T.-T.; Wu, W.-P.; Xu, C.L.; Park, K.S.; Cui, X.; Justus, S.; Lin, C.-S.; Jauregui, R.; et al. CRISPR-based genome surgery for the treatment of autosomal dominant retinitis pigmentosa. Ophthalmology 2018, 125, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xie, Y.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef]
- Xia, C.-H.; Ferguson, I.; Li, M.; Kim, A.; Onishi, A.; Li, L.; Su, B.; Gong, X. Essential function of NHE8 in mouse retina demonstrated by AAV-mediated CRISPR/Cas9 knockdown. Exp. Eye Res. 2018, 176, 29–39. [Google Scholar] [CrossRef]
- Yi, Z.; Ouyang, J.; Sun, W.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Zhang, Q. Biallelic mutations in USP45, encoding a deubiquitinating enzyme, are associated with Leber congenital amaurosis. J. Med. Genet. 2019, 56, 325–331. [Google Scholar] [CrossRef]
- McCullough, K.T.; Boye, S.L.; Fajardo, D.; Calabro, K.; Peterson, J.J.; Strang, C.E.; Chakraborty, D.; Gloskowski, S.; Haskett, S.; Samuelsson, S.; et al. Somatic Gene Editing of GUCY2D by AAV-CRISPR/Cas9 Alters Retinal Structure and Function in Mouse and Macaque. Hum. Gene Ther. 2019, 30, 571–589. [Google Scholar] [CrossRef]
- Li, F.; Hung, S.S.C.; Mohd Khalid, M.K.N.; Wang, J.-H.; Chrysostomou, V.; Wong, V.H.Y.; Singh, V.; Wing, K.; Tu, L.; Bender, J.A.; et al. Utility of Self-Destructing CRISPR/Cas Constructs for Targeted Gene Editing in the Retina. Hum. Gene Ther. 2019, 30, 1349–1360. [Google Scholar] [CrossRef]
- Yoshimi, K.; Kaneko, T.; Voigt, B.; Mashimo, T. Allele-specific genome editing and correction of disease-associated phenotypes in rats using the CRISPR–Cas platform. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N.; et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 556–563. [Google Scholar] [CrossRef]
- Feehan, J.M.; Chiu, C.N.; Stanar, P.; Tam, B.M.; Ahmed, S.N.; Moritz, O.L. Modeling Dominant and Recessive Forms of Retinitis Pigmentosa by Editing Three Rhodopsin-Encoding Genes in Xenopus Laevis Using Crispr/Cas9. Sci. Rep. 2017, 7, 6920. [Google Scholar] [CrossRef]
- Mitchell, J.; Balem, F.; Tirupula, K.; Man, D.; Dhiman, H.K.; Yanamala, N.; Ollesch, J.; Planas-Iglesias, J.; Jennings, B.J.; Gerwert, K.; et al. Comparison of the molecular properties of retinitis pigmentosa P23H and N15S amino acid replacements in rhodopsin. PLoS ONE 2019, 14, 1–14. [Google Scholar] [CrossRef]
- Wimberg, H.; Lev, D.; Yosovich, K.; Namburi, P.; Banin, E.; Sharon, D.; Koch, K.W. Photoreceptor Guanylate Cyclase (GUCY2D) Mutations Cause Retinal Dystrophies by Severe Malfunction of Ca2+-Dependent Cyclic GMP Synthesis. Front. Mol. Neurosci. 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Street, V.A.; Li, J.; Robbins, C.A.; Kallman, J.C. A DNA variant within the MYO7A promoter regulates YY1 transcription factor binding and gene expression serving as a potential dominant DFNA11 auditory genetic modifier. J. Biol. Chem. 2011, 286, 15278–15286. [Google Scholar] [CrossRef]
- Rodríguez-Pascau, L.; Coll, M.J.; Vilageliu, L.; Grinberg, D. Antisense oligonucleotide treatment for a pseudoexon-generating mutation in the NPC1 gene causing Niemann-Pick type C disease. Hum. Mutat. 2009, 30, E993–E1001. [Google Scholar] [CrossRef]
- Tan, E.; Ding, X.Q.; Saadi, A.; Agarwal, N.; Naash, M.I.; Al-Ubaidi, M.R. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 764–768. [Google Scholar] [CrossRef]
- Imamura, T.; Tsuruma, K.; Inoue, Y.; Otsuka, T.; Ohno, Y.; Ogami, S.; Yamane, S.; Shimazawa, M.; Hara, H. Involvement of cannabinoid receptor type 2 in light-induced degeneration of cells from mouse retinal cell line in vitro and mouse photoreceptors in vivo. Exp. Eye Res. 2018, 167, 44–50. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, Z.; Gao, L.; Sun, D.; Hu, X.; Xue, L.; Dai, J.; Zeng, Y.X.; Chen, S.; Pan, B.; et al. Curcumin Delays Retinal Degeneration by Regulating Microglia Activation in the Retina of rd1 Mice. Cell. Physiol. Biochem. 2017, 44, 479–493. [Google Scholar] [CrossRef]
- Balmer, J.; Zulliger, R.; Roberti, S.; Enzmann, V. Retinal cell death caused by sodium iodate involves multiple caspase-dependent and caspase-independent cell-death pathways. Int. J. Mol. Sci. 2015, 16, 15086–15103. [Google Scholar] [CrossRef] [PubMed]
- Wheway, G.; Nazlamova, L.; Turner, D.; Cross, S. 661W photoreceptor cell line as a cell model for studying retinal ciliopathies. Front. Genet. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Spalluto, C.; Wilson, D.I.; Hearn, T. Evidence for reciliation of RPE1 cells in late G1 phase, and ciliary localisation of cyclin B1. FEBS Open Bio 2013, 3, 334–340. [Google Scholar] [CrossRef]
- Di Donato, N.; Neuhann, T.; Kahlert, A.K.; Klink, B.; Hackmann, K.; Neuhann, I.; Novotna, B.; Schallner, J.; Krause, C.; Glass, I.A.; et al. Mutations in EXOSC2 are associated with a novel syndrome characterised by retinitis pigmentosa, progressive hearing loss, premature ageing, short stature, mild intellectual disability and distinctive gestalt. J. Med. Genet. 2016, 53, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Foltz, L.P.; Clegg, D.O. Patient-derived induced pluripotent stem cells for modelling genetic retinal dystrophies. Prog. Retin. Eye Res. 2019, 68, 54–66. [Google Scholar] [CrossRef]
- Bassett, A.R. Editing the genome of hiPSC with CRISPR/Cas9: Disease models. Mamm. Genome 2017, 28, 348–364. [Google Scholar] [CrossRef]
- Ovando-Roche, P.; Georgiadis, A.; Smith, A.J.; Pearson, R.A.; Ali, R.R. Harnessing the Potential of Human Pluripotent Stem Cells and Gene Editing for the Treatment of Retinal Degeneration. Curr. Stem Cell Rep. 2017, 3, 112–123. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Michaelides, M.; Holder, G.E.; Bradshaw, K.; Hunt, D.M.; Moore, A.T. Cone-rod dystrophy, intrafamilial variability, and incomplete penetrance associated with the R172W mutation in the peripherin/RDS gene. Ophthalmology 2005, 112, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Maubaret, C.G.; Vaclavik, V.; Mukhopadhyay, R.; Waseem, N.H.; Churchill, A.; Holder, G.E.; Moore, A.T.; Bhattacharya, S.S.; Webster, A.R. Autosomal Dominant Retinitis Pigmentosa with Intrafamilial Variability and Incomplete Penetrance in Two Families Carrying Mutations in PRPF8. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9304–9309. [Google Scholar] [CrossRef]
- Rose, A.M.; Bhattacharya, S.S. Variant haploinsufficiency and phenotypic non-penetrance in PRPF31-associated retinitis pigmentosa. Clin. Genet. 2016, 90, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Chapi, M.; Sabbaghi, H.; Suri, F.; Alehabib, E.; Rahimi-Aliabadi, S.; Jamali, F.; Jamshidi, J.; Emamalizadeh, B.; Darvish, H.; Mirrahimi, M.; et al. Incomplete penetrance of CRX gene for autosomal dominant form of cone-rod dystrophy. Ophthalmic Genet. 2019, 40, 259–266. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Fligor, C.M.; Langer, K.B.; Sridhar, A.; Ren, Y.; Shields, P.K.; Edler, M.C.; Ohlemacher, S.K.; Sluch, V.M.; Zack, D.J.; Zhang, C.; et al. Three-Dimensional Retinal Organoids Facilitate the Investigation of Retinal Ganglion Cell Development, Organization and Neurite Outgrowth from Human Pluripotent Stem Cells. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef]
- Wahlin, K.J.; Maruotti, J.A.; Sripathi, S.R.; Ball, J.; Angueyra, J.M.; Kim, C.; Grebe, R.; Li, W.; Jones, B.W.; Zack, D.J. Photoreceptor Outer Segment-like Structures in Long-Term 3D Retinas from Human Pluripotent Stem Cells. Sci. Rep. 2017, 7, 766. [Google Scholar] [CrossRef]
- Achberger, K.; Haderspeck, J.C.; Kleger, A.; Liebau, S. Stem cell-based retina models. Adv. Drug Deliv. Rev. 2019, 140, 33–50. [Google Scholar] [CrossRef]
- Achberger, K.; Probst, C.; Haderspeck, J.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. Elife 2019, 8. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, P.; Ma, J.H.; Cui, Z.; Yu, Q.; Liu, S.; Xue, Y.; Zhu, D.; Cao, J.; Li, Z.; et al. Modeling Retinitis Pigmentosa: Retinal Organoids Generated From the iPSCs of a Patient With the USH2A Mutation Show Early Developmental Abnormalities. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Mellough, C.B.; Collin, J.; Queen, R.; Hilgen, G.; Dorgau, B.; Zerti, D.; Felemban, M.; White, K.; Sernagor, E.; Lako, M. Systematic Comparison of Retinal Organoid Differentiation from Human Pluripotent Stem Cells Reveals Stage Specific, Cell Line, and Methodological Differences. Stem Cells Transl. Med. 2019, 8, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Peña, K.; Fontrodona, L.; Aristizábal-Corrales, D.; Torres, S.; Cornes, E.; García-Rodríguez, F.J.; Serrat, X.; González-Knowles, D.; Foissac, S.; Porta-De-La-Riva, M.; et al. Modeling of autosomal-dominant retinitis pigmentosa in Caenorhabditis elegans uncovers a nexus between global impaired functioning of certain splicing factors and cell type-specific apoptosis. RNA 2015, 21, 2119–2131. [Google Scholar] [CrossRef] [PubMed]
- Bramall, A.N.; Wright, A.F.; Jacobson, S.G.; McInnes, R.R. The Genomic, Biochemical, and Cellular Responses of the Retina in Inherited Photoreceptor Degenerations and Prospects for the Treatment of These Disorders. Annu. Rev. Neurosci. 2010, 33, 441–472. [Google Scholar] [CrossRef] [PubMed]
- Houthoofd, K.; Braeckman, B.P.; Lenaerts, I.; Brys, K.; De Vreese, A.; Van Eygen, S.; Vanfleteren, J.R. Ageing is reversed, and metabolism is reset to young levels in recovering dauer larvae of C. elegans. Exp. Gerontol. 2002, 37, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Grün, D.; Kirchner, M.; Thierfelder, N.; Stoeckius, M.; Selbach, M.; Rajewsky, N. Conservation of mRNA and protein expression during development of C.elegans. Cell Rep. 2014, 6, 565–577. [Google Scholar] [CrossRef]
- Venturini, G.; Rose, A.M.; Shah, A.Z.; Bhattacharya, S.S.; Rivolta, C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet. 2012, 8, e1003040. [Google Scholar] [CrossRef]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. DMM Dis. Models Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef]
- Colley, N.J.; Cassill, J.A.; Baker, E.K.; Zuker, C.S. Defective intracellular transport is the molecular basis of rhodopsin- dependent dominant retinal degeneration. Proc. Natl. Acad. Sci. USA 1995, 92, 3070–3074. [Google Scholar] [CrossRef]
- Wang, F.; Shi, Z.; Cui, Y.; Guo, X.; Shi, Y.B.; Chen, Y. Targeted gene disruption in Xenopus laevis using CRISPR/Cas9. Cell Biosci. 2015, 5, 1–5. [Google Scholar] [CrossRef]
- Tandon, P.; Conlon, F.; Furlow, J.D.; Horb, M.E. Expanding the genetic toolkit in Xenopus: Approaches and opportunities for human disease modeling. Dev. Biol. 2017, 426, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Feehan, J.M.; Stanar, P.; Tam, B.M.; Chiu, C.; Moritz, O.L. Generation and Analysis of Xenopus Laevis Models of Retinal Degeneration Using CRISPR/Cas9. Methods Mol. Biol. 2019, 1834, 193–207. [Google Scholar] [PubMed]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B. Genetics and early development of zebrafish. Trends Genet. 1989, 5, 283–288. [Google Scholar] [CrossRef]
- Driever, W.; Stemple, D.; Schier, A.; Solnica-Krezel, L. Zebrafish: Genetic tools for studying vertebrate development. Trends Genet. 1994, 10, 152–159. [Google Scholar] [CrossRef]
- Haffter, P.; Nüsslein-Volhard, C. Large scale genetics in a small vertebrate, the zebrafish. Int. J. Dev. Biol. 1996, 40, 221–227. [Google Scholar]
- Lawson, N.D.; Wolfe, S.A. Forward and reverse genetic approaches for the analysis of vertebrate development in the zebrafish. Dev. Cell 2011, 21, 48–64. [Google Scholar] [CrossRef]
- Meyer, A.; Schartl, M. Gene and genome duplications in vertebrates: The one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol. 1999, 11, 699–704. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Kettleborough, R.N.W.; Busch-Nentwich, E.M.; Harvey, S.A.; Dooley, C.M.; de Bruijn, E.; van Eeden, F.; Sealy, I.; White, R.J.; Herd, C.; Nijman, I.J.; et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 2013, 496, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Varshney, G.K.; Burgess, S.M. Mutagenesis and phenotyping resources in zebrafish for studying development and human disease. Brief. Funct. Genom. 2014, 13, 82–94. [Google Scholar] [CrossRef]
- Vihtelic, T.S.; Hyde, D.R. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J. Neurobiol. 2000, 44, 289–307. [Google Scholar] [PubMed]
- Fimbel, S.M.; Montgomery, J.E.; Burket, C.T.; Hyde, D.R. Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. J. Neurosci. 2007, 27, 1712–1724. [Google Scholar] [CrossRef]
- Ariga, J.; Walker, S.L.; Mumm, J.S. Multicolor Time-lapse Imaging of Transgenic Zebrafish: Visualizing Retinal Stem Cells Activated by Targeted Neuronal Cell Ablation. J. Vis. Exp. 2010, 43, 2093. [Google Scholar] [CrossRef]
- Montgomery, J.E.; Parsons, M.J.; Hyde, D.R. A Novel Model of Retinal Ablation Demonstrates That the Extent of Rod Cell Death Regulates the Origin of the Regenerated Zebrafish Rod Photoreceptors. J. Comp. Neurol. 2010, 518, 800–814. [Google Scholar] [CrossRef]
- Fraser, B.; DuVal, M.G.; Wang, H.; Allison, W.T. Regeneration of cone photoreceptors when cell ablation is primarily restricted to a particular cone subtype. PLoS ONE 2013, 8, e55410. [Google Scholar] [CrossRef]
- Tappeiner, C.; Balmer, J.; Iglicki, M.; Schuerch, K.; Jazwinska, A.; Enzmann, V.; Tschopp, M. Characteristics of rod regeneration in a novel zebrafish retinal degeneration model using N-methyl-N-nitrosourea (MNU). PLoS ONE 2013, 8, e71064. [Google Scholar] [CrossRef]
- Powell, C.; Cornblath, E.; Elsaeidi, F.; Wan, J.; Goldman, D. Zebrafish Müller glia-derived progenitors are multipotent, exhibit proliferative biases and regenerate excess neurons. Sci. Rep. 2016, 6, 24851. [Google Scholar] [CrossRef]
- Amsterdam, A.; Varshney, G.K.; Burgess, S.M. Retroviral-mediated Insertional Mutagenesis in Zebrafish. Methods Cell Biol. 2011, 104, 59–82. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient In Vivo Genome Editing Using RNA-Guided Nucleases. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Van De Weghe, J.C.; Rusterholz, T.D.S.; Latour, B.; Grout, M.E.; Aldinger, K.A.; Shaheen, R.; Dempsey, J.C.; Maddirevula, S.; Cheng, Y.-H.H.; Phelps, I.G.; et al. Mutations in ARMC9, which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish. Am. J. Hum. Genet. 2017, 101, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Vaché, C.; Besnard, T.; le Berre, P.; García-García, G.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Bolz, H.J.; Millan, J.; et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: Implications for diagnosis and therapy. Hum. Mutat. 2012, 33, 104–108. [Google Scholar] [CrossRef]
- Slijkerman, R.W.N.; Song, F.; Astuti, G.D.N.; Huynen, M.A.; van Wijk, E.; Stieger, K.; Collin, R.W.J. The pros and cons of vertebrate animal models for functional and therapeutic research on inherited retinal dystrophies. Prog. Retin. Eye Res. 2015, 48, 137–159. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Chalberg, T.W.; Schaffer, D.V. Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng. 2015, 17, 63–89. [Google Scholar] [CrossRef]
- Ramlogan-Steel, C.A.; Murali, A.; Andrzejewski, S.; Dhungel, B.; Steel, J.C.; Layton, C.J. Gene therapy and the adeno-associated virus in the treatment of genetic and acquired ophthalmic diseases in humans: Trials, future directions and safety considerations. Clin. Exp. Ophthalmol. 2019, 47, 521–536. [Google Scholar] [CrossRef]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Hum. Gene Ther. 2015, 26, 432–442. [Google Scholar] [CrossRef]
- Bowne, S.J.; Humphries, M.M.; Sullivan, L.S.; Kenna, P.F.; Tam, L.C.S.; Kiang, A.S.; Campbell, M.; Weinstock, G.M.; Koboldt, D.C.; Ding, L.; et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur. J. Hum. Genet. 2011, 19, 1074–1081. [Google Scholar] [CrossRef]
- Hull, S.; Mukherjee, R.; Holder, G.E.; Moore, A.T.; Webster, A.R. The clinical features of retinal disease due to a dominant mutation in RPE65. Mol. Vis. 2016, 22, 626–635. [Google Scholar] [PubMed]
- Perleberg, C.; Kind, A.; Schnieke, A. Genetically engineered pigs as models for human disease. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef]
- Sachs, D.H. The pig as a potential xenograft donor. Vet. Immunol. Immunopathol. 1994, 43, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.D. Nonhuman primates and translational research: Progress, opportunities, and challenges. ILAR J. 2017, 58, 141–150. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Model | Phenotype | Gene | Genomic Target | Aim | Delivery Method | Nuclease | Reference |
|---|---|---|---|---|---|---|---|
| H HeLa | adRP | RHO | p.Pro23His | KO | Lipofection (plasmid) | SpCas9 | [34] |
| H HEK293FT | LCA | CEP290 | p.Cys998* | KI | Lipofection (plasmid) | SpCas9 | [35] |
| H HEK293 | USH2/arRP | USH2A | p.Glu767Serfs*21 | KI | Lipofection (plasmid) | SpCas9 | [36] |
| p.Cys759Phe | |||||||
| M 661W | adRP | Rp9 | Exon 5 | KO | Fugene HD (plasmid) | SpCas9 | [37] |
| p.His137Leu | KI | ||||||
| H hTERT-RPE1 | xlRP | RP2 | Exon 2 | KO | Fugene HD (plasmid) | nCas9 pairs | [38] |
| RPGR | Exons 2 and 4 | KO | Undetermined | uCas9 | [39] | ||
| sarRP | PDE6D | Exon 2 | |||||
| INPP5E | Exon 1 | ||||||
| arCORD | RPGRIP1L | Exon 3 | |||||
| M NSC | arRP | Pde6b | p.Arg560Cys | C KI | Nucleofection (plasmid) | SpCas9 | [40] |
| rd12 MEFs | LCA | Rpe65 | p.Arg44* | C KI | Electroporation (RNPs) | SpCas9 | [41] |
| PD-Fibroblasts | USH2/arRP | USH2A | p.Glu767Serfs*21 | C KI | Nucleofection (RNPs) | SpCas9 | [36] |
| PD-Keratinocytes | SHRF | EXOSC2 | Exon 1 and 4 | KO | Lentiviral transduction | SpCas9 | [42] |
| PD-iPSCs | arRP | MAK | c.1513ins353 | C KI | Nucleofection (plasmid) | SpCas9 | [43] |
| LCA | CEP290 | p.Cys998* | KO | ||||
| KO | Electroporation (plasmid) | SaCas9 | |||||
| C KI | |||||||
| adRP | RHO | p.Pro23His | KO | ||||
| C KI | Undetermined (plasmid) | SpCas9 | |||||
| PRPF31 | p.Arg372Glnfs*99 | C KI | Lipofection (plasmid) | SpCas9 | [44] | ||
| Exon 7 | KO | Nucleofection (plasmid) | SpCas9 | [45] | |||
| PRPF8 | p.Pro2301Ser | C KI | Electroporation (gRNA-plasmid and Cas9 mRNA) | Cas9-Gem | [46] | ||
| xlRP | RPGR | p.His562Argfs*20 | KI | Electroporation (plasmid) | SpCas9 | [47] | |
| ESCS | NR2E3 | p.Val41Alafs*23 | KI | Lipofection (plasmid) | SpCas9 | [48] | |
| p.Arg73Ser | KI | Electroporation (plasmid) | |||||
| USH2/arRP | USH2A | p.Glu767Serfs*21 p.Cys759Phe | KI | Nucleofection (plasmid) | eSpCas9 | [49] | |
| XLRS | RS1 | p.Arg209Cys | KI | Nanodiamonds (linear DNA) | SpCas9 | [50] | |
| Caenorhabditis elegans (nematode) | adRP | prp-8 | p.Arg2310Gly | KI | Injection (RNPs) | SpCas9 | [51] |
| p.His2309del | |||||||
| snrp-200 | p.Val683Leu | ||||||
| p.Ser1087Leu | |||||||
| Danio rerio (zebrafish) | arRP | eys | p.Gly1163Valfs*14 | KO | Embryo injection (RNPs) | SpCas9 | [52] |
| pcare | p.Gly8Glu*19 | KO | Embryo injection (RNPs) | SpCas9 | [53] | ||
| adRP | rho | p.Cys322Argfs*116 | KO | Embryo injection (Cas9 mRNA and gRNAs) | SpCas9 | [54] | |
| LCA | cct2 | p.Leu394His-7del | KO | Embryo injection (RNPs) | SpCas9 | [55] | |
| USH2/arRP | ush2a | p.Cys780Glnfs*32 | KO | Embryo injection (Cas9 mRNA and gRNAs) | uCas9 | [56] | |
| p.Ala5174* | |||||||
| p.Lys2532Thrfs*56 | KI | Embryo injection (RNPs) | SpCas9 | [57] | |||
| ESCS | nr2e3 | p.Leu162Glnfs*30 | KO | Embryo injection (Cas9 mRNA and gRNAs) | uCas9 | [58] | |
| arCD | cacna2d4 | Undetermined | KO | Embryo injection (RNPs) | SpCas9 | [59] | |
| adFEVR | znf408 | p.His455Tyr | KI | Embryo injection (RNPs) | uCas9 | [60] | |
| Mus musculus (mouse) | Undetermined | Blimp1 | B108 cis-regulatory module | KO | Electroporation—subretinal injection (plasmid) | SpCas9 | [61] |
| LCA | Cep290 | p.Cys998* | KO | AAV transduction (subretinal injection) | SpCas9 | [35] | |
| Exon 3 | KO | AAV transduction (subretinal injection) | SpCas9 | [62] | |||
| Rpe65 | p.Asp477Gly | KI | Embryo injection (Cas9 mRNA and gRNAs) | SpCas9 | [63] | ||
| p.Arg44* | C KI | AAV transduction (subretinal injection) | SpCas9 | [41] | |||
| Kcnj13 | Exon 2 | KO | Zygote injection (Cas9 mRNA and gRNAs) | SpCas9 | [64] | ||
| OCA1 | Tyr | 5’ region | KO | Zygote injection (Cas9 mRNA and gRNAs) | SpCas9 | [65] | |
| Thy1-YFP | YFP | 5’ region | KO | AAV transduction (intravitreal injection) | SpCas9 | [66] | |
| arRP | Pde6b | p.Arg560Cys | KI | Electroporation—subretinal injection (plasmid) | SpCas9 | [40] | |
| p.Tyr347Ter | C KI | Embryo injection (gRNA-plasmid and Cas9 protein) | SpCas9 | [67] | |||
| Reep6 | p.Leu135Pro | KI | Embryo injection (Cas9 mRNA and gRNAs) | SpCas9 | [68] | ||
| Exon 4 | KO | Embryo injection (Cas9 mRNA and gRNAs) | SpCas9 | [69] | |||
| Arl2bp | Exon 2 | KO | Embryo injection (Cas9 mRNA and gRNAs) | SpCas9 | [70] | ||
| Hkdc1 | Exon 2 | KO | Undetermined (plasmid) | uCas9 | [71] | ||
| adRP | RHO | p.Pro23His | KO | Electroporation (plasmid) | SpCas9 | [34] | |
| Electroporation (plasmid) and AAV transduction (intravitreal injection) | SaCas9 and SaCas9-KKH | [72] | |||||
| Rho/RHO | Exon 1 | KO | AAV transduction (subretinal injection) | SpCas9 | [73] | ||
| arRP/sarRP | Cwc27 | p.Lys338Glyfs*25 | KO | Embryo injection (Cas9 mRNA and gRNAs) | SpCas9 | [74] | |
| arRP/adRP | Nrl | Undetermined | KO | AAV transduction (subretinal injection) | SpCas9 | [75] | |
| [76] | |||||||
| RD | Slc9a8 | Promoter | KO | AAV transduction (subretinal injection) | nmCas9 | [77] | |
| Usp45 | Exon 14 | KO | Embryo injection (Cas9 mRNA and gRNAs) | uCas9 | [78] | ||
| adCORD | Gucy2e | Exon 2 and 4 | KO | AAV transduction (subretinal injection) | SaCas9 | [79] | |
| Thy1-YFP | YFP | Undetermined | KO | AAV transduction (subretinal injection | SpCas9 | [80] | |
| XLRS | Rs1 | p.Arg209Cys | KI | Nanodiamonds—intravitreal injection (linear DNA) | SpCas9 | [50] | |
| Rattus norvegicus (rat) | OCA1 | Tyr | Exon 2 | KO | Embryo injection (Cas9 mRNA and gRNAs) | uCas9 | [81] |
| p.Arg299His | KI | ||||||
| adRP | Rho | p.Ser334Ter | KO | Electroporation—subretinal injection (plasmid) | SpCas9 | [82] | |
| Xenopus laevis (frog) | adRP | rho | Exon1 | KO | Embryo injection (Cas9 mRNA and gRNAs) | SpCas9 | [83] |
| Exon 5 | KI | ||||||
| Drosophila melanogaster (fly) | SHRF | rrp4 | Exon 1 and 4 | KO | Embryo injection (gRNA-plasmid into Cas9-expressing strain) | SpCas9 | [42] |
| Sus scrofa domesticus (pig) | adRP | RHO | p.Pro23His | KO | AAV transduction (subretinal injection) | SaCas9 | [43] |
| Macaca fascicularis (macaque) | adCORD | GUCY2D | Exon 2 and 4 | KO | AAV transduction (subretinal injection) | saCas9 | [79] |
| Method | Advantages | Disadvantages |
|---|---|---|
| Microinjection | Liberated right into the cell High efficacy | Time-consuming Technique expertise |
| Electroporation | Normalized open-access protocols High effectiveness with plasmids | In vitro and ex vivo cell restriction Cell cytotoxicity Not all cells are susceptible |
| Lipofection | Works in many cell types Easy manipulation Inexpensive Reduced off-targets | Exclusive for cell culture Lysosome degradation |
| Nanodiamonds | Highly efficient delivery High biocompatibility Water solubility Fully accessible surface Inexpensive | Genotoxicity High pressure and temperature for synthesis Tissue distribution problems |
| AAVs | Low immunogenicity and cytotoxicity Reduced off-targets High efficacy Low immune response detected Infect both dividing and non-dividing cells | Limited cargo capacity (3.5–4 kb) High cost Technique expertise Safety obstacle Not easy to scale-up |
| Lentivirus | Expression stability Can be applied in a broad range of cell types Higher efficacy if constructs are shortened Low immune response detected Infect both dividing and non-dividing cells | Limited cargo capacity (8–9 kb) Arbitrary integration Technique expertise Safety obstacle Not easy to scale-up |
| Microinjection delivery is based on the use of a 0.5–5.0 µm diameter needle to deliver components into a cell or intercellular space, in this case the Cas9 protein and sgRNAs in any form. The electroporation method requires high voltage currents with the purpose of opening nanopores in the cellular membrane to inlet the CRISPR components resuspended in a specific buffer. Nucleofection is a specific electroporation-based method that allows the direct entry of the components into the nucleus. Lipofection consists in the introduction of the DNA components via a liposome-based transfection, in which synthetic cationic lipids aggregate around the negatively-charged DNA molecules. Cellular uptake is based on the fusion of these liposome-like structures with the phospholipidic membrane. Nanodiamonds are carbon nanomaterials which are suspended in a colloidal solution that allow the binding with or coating of biological material for cell transfection, mainly penetrating by the clathrin-mediated endocytosis pathway. The viral transduction method leverages the natural potential of viruses to infect cells, where the vectors have been deprived of the essential pathogenic genes in their replication. The most commonly used are AAVs and lentivirus. AAVs, which consist of single-stranded DNA, present several serotype versions (allowing tissue-specific transduction) and are considered a safe option, given that they are not associated to human diseases showing low immunogenicity and entailing low cellular toxicity (as they do not integrate into the host genome). Lentiviruses are retroviruses derived from a provirus of HIV that proffer stable expression in both dividing and post-mitotic cells due to their host genome integration, and that additionally accommodate cargos up to 5–6 kb in size. | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Millán, J.M.; García-García, G. Application of CRISPR Tools for Variant Interpretation and Disease Modeling in Inherited Retinal Dystrophies. Genes 2020, 11, 473. https://doi.org/10.3390/genes11050473
Fuster-García C, García-Bohórquez B, Rodríguez-Muñoz A, Millán JM, García-García G. Application of CRISPR Tools for Variant Interpretation and Disease Modeling in Inherited Retinal Dystrophies. Genes. 2020; 11(5):473. https://doi.org/10.3390/genes11050473
Chicago/Turabian StyleFuster-García, Carla, Belén García-Bohórquez, Ana Rodríguez-Muñoz, José M. Millán, and Gema García-García. 2020. "Application of CRISPR Tools for Variant Interpretation and Disease Modeling in Inherited Retinal Dystrophies" Genes 11, no. 5: 473. https://doi.org/10.3390/genes11050473
APA StyleFuster-García, C., García-Bohórquez, B., Rodríguez-Muñoz, A., Millán, J. M., & García-García, G. (2020). Application of CRISPR Tools for Variant Interpretation and Disease Modeling in Inherited Retinal Dystrophies. Genes, 11(5), 473. https://doi.org/10.3390/genes11050473