Pharmacogenomic Markers of Methotrexate Response in the Consolidation Phase of Pediatric Acute Lymphoblastic Leukemia Treatment

 , , ,
, , ,
Abstract
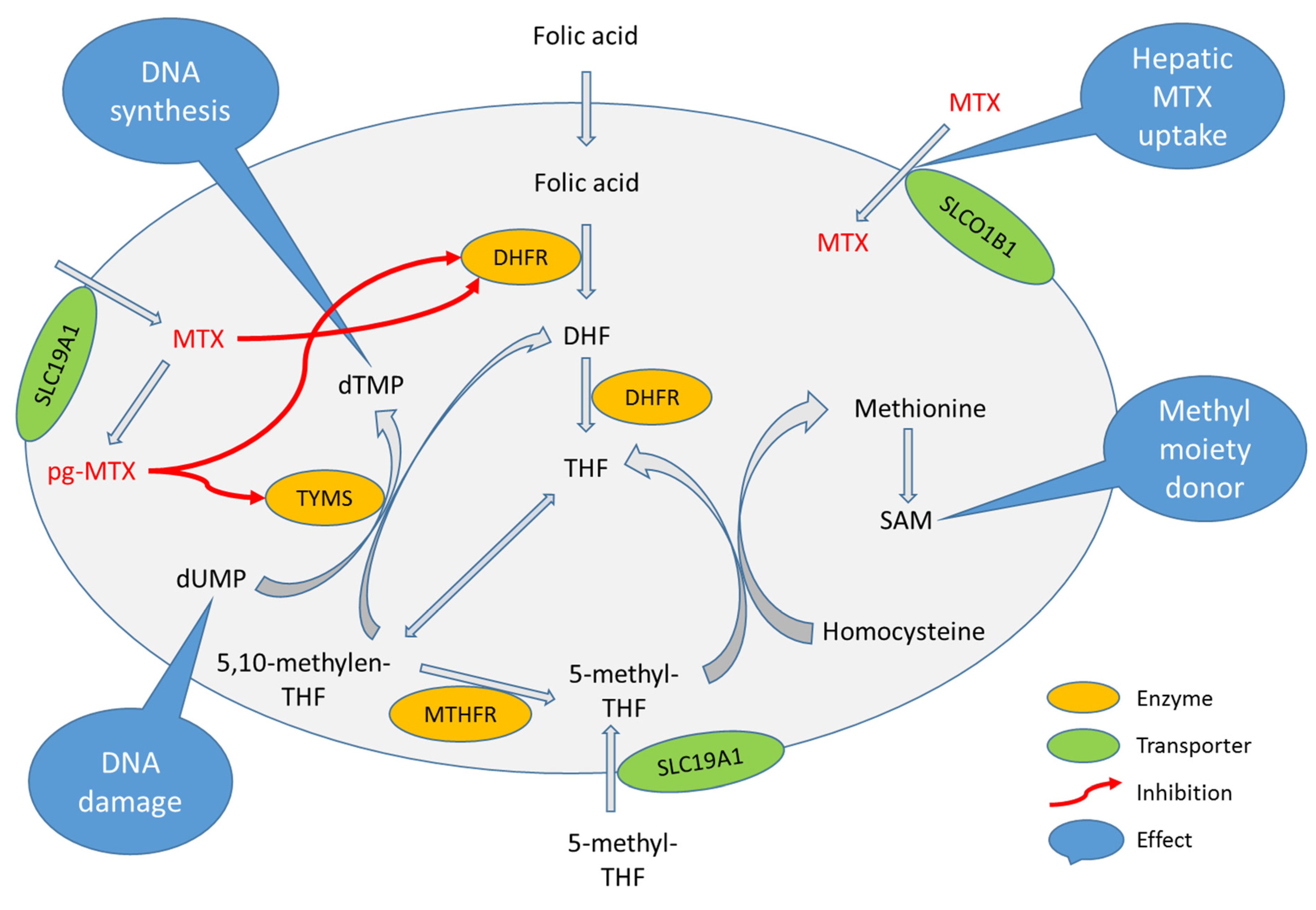
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Pharmacokinetics of MTX in the Consolidation Therapy Phase
2.3. MTX Toxicity in the Consolidation Therapy Phase
2.4. Genetic Variants Detection
2.5. Statistical Analysis
3. Results
3.1. Population PGx
3.2. Patients
3.3. MTX Kinetics during Consolidation Therapy Phase
3.4. MTX Toxicity during Consolidation Therapy Phase
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Schrappe, M.; Reiter, A.; Ludwig, W.-D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: Results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 2000, 95, 3310–3322. [Google Scholar] [PubMed]
- Pavlovic, S.; Kotur, N.; Stankovic, B.; Zukic, B.; Gasic, V.; Dokmanovic, L. Pharmacogenomic and Pharmacotranscriptomic Profiling of Childhood Acute Lymphoblastic Leukemia: Paving the Way to Personalized Treatment. Genes 2019, 10, 191. [Google Scholar] [CrossRef]
- ALL IC-BFM 2009. A Randomized Trial of the I-BFM-SG for the Management of Childhood non-B Acute Lymphoblastic Leukemia Final Version of Therapy Protocol from August-14-2009. Available online: http://www.bialaczka.org/wp-content/uploads/2016/10/ALLIC_BFM_2009.pdf (accessed on 28 February 2020).
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid (Aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Martin-Guerrero, I.; Ballesteros, J.; Piñán, M.; Garcia-Miguel, P.; Navajas, A.; Garcia-Orad, A. Polymorphisms of the SLCO1B1 Gene Predict Methotrexate-Related Toxicity in Childhood Acute Lymphoblastic Leukemia. Pediatr. Blood Cancer 2011, 57, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Gutierrez-Camino, A.; Bilbao-Aldaiturriaga, N.; Pombar-Gomez, M.; Martin-Guerrero, I.; Garcia-Orad, A. Pharmacogenetics of childhood acute lymphoblastic leukemia. Pharmacogenomics 2014, 15, 1383–1398. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y. The role of multidrug resistance efflux transporters in antifolate resistance and folate homeostasis. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2006, 9, 227–246. [Google Scholar] [CrossRef]
- Wang, L.; Weinshilboum, R. Thiopurine S-methyltransferase pharmacogenetics: Insights, challenges and future directions. Oncogene 2006, 25, 1629–1638. [Google Scholar] [CrossRef]
- Rudin, S.; Marable, M.; Huang, R. The Promise of Pharmacogenomics in Reducing Toxicity During Acute Lymphoblastic Leukemia Maintenance Treatment. Genom. Proteom. Bioinform. 2017, 15, 82–93. [Google Scholar] [CrossRef]
- Ceppi, F.; Gagné, V.; Douyon, L.; Quintin, C.; Colombini, A.; Parasole, R.; Buldini, B.; Basso, G.; Conter, V.; Cazzaniga, G.; et al. DNA variants in DHFR gene and response to treatment in children with childhood B ALL: Revisited in AIEOP-BFM protocol. Pharmacogenomics 2018, 19, 105–112. [Google Scholar] [CrossRef]
- Dulucq, S.; St-Onge, G.; Gagne, V.; Ansari, M.; Sinnett, D.; Labuda, D.; Moghrabi, A.; Krajinovic, M. DNA variants in the dihydrofolate reductase gene and outcome in childhood ALL. Blood 2008, 111, 3692–3700. [Google Scholar] [CrossRef]
- Krajinovic, M.; Costea, I.; Primeau, M.; Dulucq, S.; Moghrabi, A. Combining several polymorphisms of thymidylate synthase gene for pharmacogenetic analysis. Pharmacogenom. J. 2005, 5, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-G.; Gao, C.; Zhang, R.-D.; Zhao, X.-X.; Cui, L.; Li, W.-J.; Chen, Z.-P.; Yue, Z.-X.; Zhang, Y.-Y.; Wu, M.-Y.; et al. Polymorphisms in methotrexate transporters and their relationship to plasma methotrexate levels, toxicity of high-dose methotrexate, and outcome of pediatric acute lymphoblastic leukemia. Oncotarget 2017, 8, 37761–37772. [Google Scholar] [CrossRef]
- Sepe, D.; McWilliams, T.; Chen, J.; Kershenbaum, A.; Zhao, H.; La, M.; Devidas, M.; Lange, B.; Rebbeck, T.; Aplenc, R. Germline genetic variation and treatment response on CCG-1891. Pediatr. Blood Cancer 2012, 58, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Radtke, S.; Zolk, O.; Renner, B.; Paulides, M.; Zimmermann, M.; Moricke, A.; Stanulla, M.; Schrappe, M.; Langer, T. Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 2013, 121, 5145–5153. [Google Scholar] [CrossRef]
- Ramsey, L.; Panetta, J.; Smith, C.; Yang, W.; Fan, Y.; Winick, N.; Martin, P.; Cheng, C.; Devidas, M.; Pui, C.-H.; et al. Genome-wide study of methotrexate clearance replicates SLCO1B1. Blood 2012, 121. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Bruun, G.H.; Yang, W.; Trevino, L.R.; Vattathil, S.; Scheet, P.; Cheng, C.; Rosner, G.L.; Giacomini, K.M.; Fan, Y.; et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Trevino, L.R.; Shimasaki, N.; Yang, W.; Panetta, J.C.; Cheng, C.; Pei, D.; Chan, D.; Sparreboom, A.; Giacomini, K.M.; Pui, C.H.; et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5972–5978. [Google Scholar] [CrossRef] [PubMed]
- Zaïr, Z.M.; Singer, D.R.J. Influx transporter variants as predictors of cancer chemotherapy-induced toxicity: Systematic review and meta-analysis. Pharmacogenomics 2016, 17, 1189–1205. [Google Scholar] [CrossRef]
- He, H.R.; Liu, P.; He, G.H.; Dong, W.H.; Wang, M.Y.; Dong, Y.L.; Lu, J. Association between reduced folate carrier G80A polymorphism and methotrexate toxicity in childhood acute lymphoblastic leukemia: A meta-analysis. Leuk. Lymphoma 2014, 55, 2793–2800. [Google Scholar] [CrossRef]
- Lopez-Lopez, E.; Martin-Guerrero, I.; Ballesteros, J.; Garcia-Orad, A. A systematic review and meta-analysis of MTHFR polymorphisms in methotrexate toxicity prediction in pediatric acute lymphoblastic leukemia. Pharmacogenom. J. 2013, 13, 498–506. [Google Scholar] [CrossRef]
- Oosterom, N.; Berrevoets, M.; den Hoed, M.A.H.; Zolk, O.; Hoerning, S.; Pluijm, S.M.F.; Pieters, R.; de Jonge, R.; Tissing, W.J.E.; van den Heuvel-Eibrink, M.M.; et al. The role of genetic polymorphisms in the thymidylate synthase (TYMS) gene in methotrexate-induced oral mucositis in children with acute lymphoblastic leukemia. Pharmacogenet. Genom. 2018, 28, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Liu, Y.W.; Wang, S.Z.; Li, X.L.; Nie, X.L.; Yu, X.T.; Zhao, L.B.; Wang, X.L. Associations between the C677T and A1298C polymorphisms of MTHFR and the toxicity of methotrexate in childhood malignancies: A meta-analysis. Pharmacogenom. J. 2018, 18, 450–459. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, j.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed]
- Carmona, B.; Guerreiro, C.; Cravo, M.; Nobre-Leitao, C.; Brito, M. 5′ and 3′ UTR thymidylate synthase polymorphisms modulate the risk of colorectal cancer independently of the intake of methyl group donors. Mol. Med. Rep. 2008, 1, 747–752. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graziano, F.; Ruzzo, A.; Loupakis, F.; Santini, D.; Catalano, V.; Canestrari, E.; Maltese, P.; Bisonni, R.; Fornaro, L.; Baldi, G.; et al. Liver-only metastatic colorectal cancer patients and thymidylate synthase polymorphisms for predicting response to 5-fluorouracil-based chemotherapy. Br. J. Cancer 2008, 99, 716–721. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hubner, R.; Liu, J.-F.; Sellick, G.; Logan, R.; Houlston, R.; Muir, K. Thymidylate synthase polymorphisms, folate and B-vitamin intake, and risk of colorectal adenoma. Br. J. Cancer 2007, 97, 1449–1456. [Google Scholar] [CrossRef][Green Version]
- Shi, Q.; Zhang, Z.; Neumann, A.; Li, G.; Spitz, M.; Wei, Q. Case–control analysis of thymidylate synthase polymorphism and risk of lung cancer. Carcinogenesis 2005, 26, 649–656. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, Q.; Sturgis, E.; Spitz, M.; Hong, W.; Wei, Q. Thymidylate synthase 5′- and 3′-untranslated region polymorphisms associated with risk and progression of squamous cell carcinoma of the head and neck. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 10, 7903–7910. [Google Scholar] [CrossRef][Green Version]
- Cheok, M.; Evans, W. Acute lymphoblastic leukemia: A model for the pharmacogenomics of cancer therapy. Nat. Rev. Cancer 2006, 6, 117–129. [Google Scholar] [CrossRef]
- Hou, Z.; Matherly, L. Biology of the major facilitative folate transporters SLC19A1 and SLC46A1. Curr. Top. Membr. 2014, 73, 175–204. [Google Scholar] [CrossRef]
- Matherly, L.; Hou, Z.; Deng, Y. Human reduced folate carrier: Translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007, 26, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Rady, P.L.; Szucs, S.; Matalon, R.K.; Grady, J.; Hudnall, S.D.; Kellner, L.H.; Nitowsky, H. Genetic Polymorphism (G80A) of Reduced Folate Carrier Gene in Ethnic Populations. Mol. Genet. Metab. 2001, 73, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Drori, S.; Jansen, G.; Mauritz, R.; Peters, G.; Assaraf, Y. Clustering of Mutations in the First Transmembrane Domain of the Human Reduced Folate Carrier in GW1843U89-resistant Leukemia Cells with Impaired Antifolate Transport and Augmented Folate Uptake. J. Biol. Chem. 2000, 275, 30855–30863. [Google Scholar] [CrossRef] [PubMed]
- Gifford, A.; Haber, M.; Witt, T.; Whetstine, J.; Taub, J.; Matherly, L.; Norris, M. Role of the E45K-reduced folate carrier gene mutation in methotrexate resistance in human leukemia cells. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund U.K 2003, 16, 2379–2387. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jansen, G.; Mauritz, R.; Drori, S.; Sprecher, H.; Kathmann, I.; Bunni, M.; Priest, D.G.; Noordhuis, P.; Schornagel, J.H.; Pinedo, H.M.; et al. A Structurally Altered Human Reduced Folate Carrier with Increased Folic Acid Transport Mediates a Novel Mechanism of Antifolate Resistance. J. Biol. Chem. 1998, 273, 30189–30198. [Google Scholar] [CrossRef]
- Tse, A.; Brigle, K.; Taylor, S.M.; Moran, R.G. Mutations in the Reduced Folate Carrier Gene Which Confer Dominant Resistance to 5,10-Dideazatetrahydrofolate. J. Biol. Chem. 1998, 273, 25953–25960. [Google Scholar] [CrossRef]
- Zhao, R.; Assaraf, Y.; Goldman, I. A Mutated Murine Reduced Folate Carrier (RFC1) with Increased Affinity for Folic Acid, Decreased Affinity for Methotrexate, and an Obligatory Anion Requirement for Transport Function. J. Biol. Chem. 1998, 273, 19065–19071. [Google Scholar] [CrossRef]
- Zhao, R.; Assaraf, Y.; Goldman, I.D. A reduced folate carrier mutation produces substrate-dependent alterations in carrier mobility in murine leukemia cells and methotrexate resistance with conservation of growth in 5-formyltetrahydrofolate. J. Biol. Chem. 1998, 273, 7873–7879. [Google Scholar] [CrossRef]
- Zhao, R.; Sharina, I.; Goldman, I. Pattern of Mutations that Results in Loss of Reduced Folate Carrier Function under Antifolate Selective Pressure Augmented by Chemical Mutagenesis. Mol. Pharmacol. 1999, 56, 68–76. [Google Scholar] [CrossRef]
- Whetstine, J.R.; Gifford, A.J.; Witt, T.; Liu, X.Y.; Flatley, R.M.; Norris, M.; Haber, M.; Taub, J.W.; Ravindranath, Y.; Matherly, L.H. Single Nucleotide Polymorphisms in the Human Reduced Folate Carrier. Clin. Cancer Res. 2001, 7, 3416–3422. [Google Scholar]
- Galbiatti-Dias, A.L.; Ruiz, M.; Pinto, D.; Raposo, L.; Maníglia, J.; Pavarino, É.; Goloni-Bertollo, E. A80G polymorphism of reduced folate carrier 1 (RFC1) gene and head and neck squamous cell carcinoma etiology in Brazilian population. Mol. Biol. Rep. 2011, 38, 1071–1078. [Google Scholar] [CrossRef]
- Leyva-Vazquez, M.; Organista-Nava, J.; Gómez-Gómez, Y.; Contreras-Quiroz, A.; Flores-Alfaro, E.; Illades-Aguiar, B. Polymorphism G80A in the Reduced Folate Carrier Gene and its Relationship to Survival and Risk of Relapse in Acute Lymphoblastic Leukemia. J. Investig. Med. 2012, 60, 1064–1067. [Google Scholar] [CrossRef]
- Jonge, R.; Tissing, W.; Hooijberg, J.; Jansen, G.; Kaspers, G.; Lindemans, J.; Peters, G.; Pieters, R. Polymorphisms in folate-related genes and risk of pediatric acute lymphoblastic leukemia. Blood 2008, 113, 2284–2289. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Fontes, A.; Silva, K.; Sant’Ana, T.; Ramos, F.; Marques-Salles, T.; Pombo-de-Oliveira, M.; Muniz, M. Polymorphisms Involved in Folate Metabolism Pathways and the Risk of the Development of Childhood Acute Leukemia. Genet. Test. Mol. Biomark. 2013, 17, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Gregers, J.; Christensen, I.; Dalhoff, K.; Lausen, B.; Schroeder, H.; Rosthøj, S.; Carlsen, N.; Schmiegelow, K.; Peterson, C. The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 2010, 115, 4671–4677. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giletti, A.; Esperon, P. Genetic markers in methotrexate treatments. Pharmacogenom. J. 2018, 18, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Csordas, K.; Lautner-Csorba, O.; Semsei, A.; Harnos, A.; Md, M.; Erdélyi, D.; Eipel, O.; Szalai, C.; Kovacs, G. Associations of novel genetic variations in the folate-related and ARID5B genes with the pharmacokinetics and toxicity of high-dose methotrexate in paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2014, 166, 410–420. [Google Scholar] [CrossRef]
- Fukushima, H.; Fukushima, T.; Sakai, A.; Suzuki, R.; Nakajima-Yamaguchi, R.; Kobayashi, C.; Iwabuchi, A.; Saito, M.; Yoshimi, A.; Nakao, T.; et al. Polymorphisms of MTHFR Associated with Higher Relapse/Death Ratio and Delayed Weekly MTX Administration in Pediatric Lymphoid Malignancies. Leuk. Res. Treat. 2013, 2013, 238528. [Google Scholar] [CrossRef]
- Giletti, A.; Vital, M.; Lorenzo, M.; Cardozo, P.; Borelli, G.; Gabus, R.; Martínez, L.; Díaz, L.; Assar, R.; Rodriguez, M.N.; et al. Methotrexate pharmacogenetics in Uruguayan adults with hematological malignant diseases. Eur. J. Pharm. Sci. 2017, 109, 480–485. [Google Scholar] [CrossRef]
- Lopez-Lopez, E.; Ballesteros, J.; Piñán, M.; Toledo, J.; García de Andoin, N.; Garcia-Miguel, P.; Navajas, A.; Garcia-Orad, A. Polymorphisms in the methotrexate transport pathway: A new tool for MTX plasma level prediction in pediatric acute lymphoblastic leukemia. Pharmacogenet. Genom. 2013, 23, 53–61. [Google Scholar] [CrossRef]
- Lima, A.; Azevedo, R.; Sousa, H.; Seabra, V.; Medeiros, R. Current approaches for TYMS polymorphisms and their importance in molecular epidemiology and pharmacogenetics. Pharmacogenomics 2013, 14, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Lurje, G.; Manegold, P.; Ning, Y.; Pohl, A.; Zhang, W.; Lenz, H.-J. Thymidylate synthase gene variations: Predictive and prognostic markers. Mol. Cancer Ther. 2009, 8, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Scofield, D.; Hong, X. The Evolution of Transcription-Initiation Sites. Mol. Biol. Evol. 2005, 22, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Salonga, D.; Park, J.M.; Danenberg, K.D.; Uetake, H.; Brabender, J.; Omura, K.; Watanabe, G.; Danenberg, P.V. Different Lengths of a Polymorphic Repeat Sequence in the Thymidylate Synthase Gene Affect Translational Efficiency but Not Its Gene Expression. Clin. Cancer Res. 2001, 7, 4096–4101. [Google Scholar] [PubMed]
- Lima, A.; Seabra, V.; Bernardes, M.; Azevedo, R.; Sousa, H.; Medeiros, R. Role of Key TYMS Polymorphisms on Methotrexate Therapeutic Outcome in Portuguese Rheumatoid Arthritis Patients. PLoS ONE 2014, 9, e108165. [Google Scholar] [CrossRef] [PubMed]
- Pullmann, R.; Abdelmohsen, K.; Lal, A.; Martindale, J.L.; Ladner, R.D.; Gorospe, M. Differential Stability of Thymidylate Synthase 3′-Untranslated Region Polymorphic Variants Regulated by AUF1. J. Biol. Chem. 2006, 281, 23456–23463. [Google Scholar]
- Allegra, C.J.; Chabner, B.A.; Drake, J.C.; Lutz, R.; Rodbard, D.; Jolivet, J. Enhanced inhibition of thymidylate synthase by methotrexate polyglutamates. J. Biol. Chem. 1985, 260, 9720–9726. [Google Scholar]
- Tsukada, T.; Nakano, T.; Miyata, T.; Sasaki, S. Life-Threatening Gastrointestinal Mucosal Necrosis during Methotrexate Treatment for Rheumatoid Arthritis. Case Rep. Gastroenterol. 2013, 7, 470–475. [Google Scholar] [CrossRef]
- Pullarkat, S.; Stoehlmacher, J.; Ghaderi, V.; Xiong, Y.P.; Ingles, S.; Sherrod, A.; Warren, R.; Tsao-Wei, D.; Groshen, S.; Lenz, H. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenom. J. 2001, 1, 65–70. [Google Scholar] [CrossRef]
- Wang, B.; Walsh, S.; Saif, W. Pancytopenia and Severe Gastrointestinal Toxicities Associated with 5-Fluorouracil in a Patient with Thymidylate Synthase (TYMS) Polymorphism. Cureus 2016, 8, e798. [Google Scholar] [CrossRef]
- Kishi, S.; Cheng, C.; French, D.; Pei, D.; Das, S.; Cook, E.; Hijiya, N.; Rizzari, C.; Rosner, G.; Frudakis, T.; et al. Ancestry and pharmacogenetics of antileukemic dug toxicity. Blood 2007, 109, 4151–4157. [Google Scholar] [CrossRef] [PubMed]
- Mizzi, C.; Dalabira, E.; Kumuthini, J.; Dzimiri, N.; Balogh, I.; Basak, A.N.; Böhm, R.; Borg, J.; Borgiani, P.; Božina, N.; et al. A European Spectrum of Pharmacogenomic Biomarkers: Implications for Clinical Pharmacogenomics. PLoS ONE 2016, 11, e0162866. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Sun, L.-L.; Zeng, W.-X.; Wu, W.-S.; Zhang, G.-L. Effects of a microRNA binding site polymorphism in SLC19A1 on methotrexate concentrations in Chinese children with acute lymphoblastic leukemia. Med. Oncol. (Northwood Lond. Engl.) 2014, 31, 62. [Google Scholar] [CrossRef] [PubMed]
- Nacional Cancer Institute. Cancer Therapy Evaluation Program: Common Toxicity Criteria Manual; National Cancer Institute: Bethesda, MD, USA, 1999; pp. 1–29. [Google Scholar]
- BFM International Trials. Available online: http://www.bfm-international.org/trials.php (accessed on 28 February 2020).
- Neidhart, M. DNA Methylation and Complex Human Disease, 1st ed.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Relton, C.; Wilding, C.; Pearce, M.; Laffling, A.J.; Jonas, P.A.; Lynch, S.A.; Tawn, E.; Burn, J. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J. Med. Genet. 2004, 41, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Zinck, J.; MacFarlane, A. Approaches for the Identification of Genetic Modifiers of Nutrient Dependent Phenotypes: Examples from Folate. Front. Nutr. 2014, 1, 8. [Google Scholar] [CrossRef]
- Amos, W.; Driscoll, E.; Hoffman, J. Candidate genes versus genome-wide associations: Which are better for detecting genetic susceptibility to infectious disease? Proc. Biol. Sci. R. Soc. 2010, 278, 1183–1188. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genetic Variants | Genotype | Serbian Childhood ALL Patients | Serbian Control Group | European Caucasian Population | p Value b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | HW | MAF (%) | N | % | HW | MAF (%) | N | % | MAF (%) | |||
| TYMSa rs34743033 28bp VNTR | 3R/3R | 49 | 33.1 | 0.047 | 45.7 | 33 | 31.7 | 0.81 | 42.8 | 981 | 29.9 | 46.3 | 0.39 |
| 3R/2R | 61 | 41.2 | 51 | 49.0 | 1566 | 47.7 | |||||||
| 2R/2R | 37 | 25.0 | 19 | 18.3 | 735 | 22.4 | |||||||
| 3R/4R | 1 | 0.7 | NA | NA | 1 | 1.0 | NA | NA | NA | NA | NA | NA | |
| TYMS rs34489327 6bp indel | Ins/Ins | 70 | 47.3 | 0.57 | 33.1 | 47 | 45.2 | 0.92 | 33.2 | 1532 | 46.3 | 32.8 | 0.92 |
| Ins/Del | 61 | 41.2 | 45 | 43.3 | 1386 | 41.8 | |||||||
| Del/Del | 17 | 11.5 | 12 | 11.5 | 394 | 11.9 | |||||||
| MTHFR rs1801133 c.677 C>T | C/C | 70 | 47.3 | 0.57 | 32.1 | 113 | 47.5 | 0.013 | 33.8 | 204 | 40.6 | 36.5 | 0.32 |
| C/T | 61 | 41.2 | 89 | 37.4 | 231 | 45.9 | |||||||
| T/T | 17 | 11.5 | 36 | 15.1 | 68 | 13.5 | |||||||
| MTHFR rs1801131 c.1298 A>C | A/A | 71 | 48.6 | 0.252 | 31.4 | 115 | 48.3 | 0.761 | 30.9 | 239 | 47.5 | 31.3 | 0.86 |
| A/C | 57 | 39.0 | 99 | 41.6 | 213 | 42.3 | |||||||
| C/C | 18 | 12.3 | 24 | 10.1 | 51 | 10.1 | |||||||
| DHFR rs442767 -680 C>A | C/C | 63 | 44.7 | 1 | 31.4 | 45 | 43.3 | 0.86 | 34.6 | 232 | 46.1 | 31.9 | 0.45 |
| C/A | 63 | 44.7 | 46 | 44.2 | 221 | 43.9 | |||||||
| A/A | 15 | 10.6 | 13 | 12.5 | 50 | 9.9 | |||||||
| DHFR rs1643641 -675 A>G | A/A | 74 | 52.5 | 0.31 | 27.4 | 55 | 52.9 | 0.31 | 26.0 | 286 | 56.9 | 25.2 | 0.82 |
| A/G | 53 | 37.6 | 44 | 42.3 | 180 | 35.8 | |||||||
| G/G | 14 | 9.9 | 5 | 4.8 | 37 | 7.4 | |||||||
| DHFR rs1650695 -556 T>C | T/T | 73 | 51.8 | 0.54 | 27.4 | 55 | 52.9 | 0.52 | 26.4 | 285 | 56.7 | 25.3 | 0.74 |
| T/C | 55 | 39.0 | 43 | 41.3 | 181 | 36.0 | |||||||
| C/C | 13 | 9.2 | 6 | 5.8 | 37 | 7.4 | |||||||
| DHFR rs1650696 -464 A>T | A/A | 72 | 51.1 | 0.68 | 27.7 | 55 | 52.9 | 0.52 | 26.4 | 286 | 56.9 | 25.2 | 0.72 |
| A/T | 56 | 39.7 | 43 | 41.3 | 180 | 35.8 | |||||||
| T/T | 13 | 9.2 | 6 | 5.8 | 37 | 7.4 | |||||||
| DHFR rs408626 -317 A>G | A/A | 49 | 34.8 | 0.60 | 28.2 | 37 | 35.6 | 0.99 | 40.4 | 150 | 29.8 | 45.0 | 0.22 |
| A/G | 71 | 50.4 | 50 | 48.1 | 253 | 50.3 | |||||||
| G/G | 21 | 14.9 | 17 | 16.3 | 100 | 19.9 | |||||||
| SLC19A1 rs1051266 c.80 G>A | G/G | 39 | 26.9 | 0.40 | 49.0 | 72 | 30.3 | 0.80 | 65.7 | 158 | 31.4 | 45.1 | 0.92 |
| G/A | 67 | 46.2 | 116 | 48.7 | 236 | 46.9 | |||||||
| A/A | 39 | 26.9 | 50 | 21.0 | 109 | 21.7 | |||||||
| SLCO1B1 rs4149056 c.521 T>C | T/T | 100 | 71.9 | 0.06 | 15.5 | 100 | 74.6 | 1 | 13.4 | 351 | 69.8 | 16.1 | 0.28 |
| T/C | 32 | 23.0 | 32 | 23.9 | 142 | 28.2 | |||||||
| C/C | 7 | 5.0 | 2 | 1.5 | 10 | 2.0 | |||||||
| mM Protocol (MD-MTX), n = 102 Toxicity Grade | M Protocol (HD-MTX), n = 28 Toxicity Grade | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | |
| GIT toxicity | 61 | 17 | 22 | 2 | - | 15 | 3 | 9 | 1 | - |
| Oral mucositis | 75 | 19 | 6 | 2 | - | 19 | 4 | 5 | - | - |
| Liver toxicity | 81 | 16 | 4 | 1 | - | 21 | 4 | 1 | 1 | 1 |
| Nephrotoxicity | 99 | 3 | - | - | - | 23 | 4 | 1 | - | - |
| Skin toxicity | 102 | - | - | - | - | 26 | - | 2 | - | - |
| Neurotoxicity | 96 | 3 | 3 | - | - | 26 | 1 | 1 | - | - |
| Gene | Variant | dbSNP | Alleles | MAF | Oral Mucositis | GIT toxicity | Hepatotoxicity | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | p | OR | 95% CI | p | OR | 95% CI | p | |||||
| MTHFR | c.677 C>T | rs1801133 | C/T | 0.32 | 1.54 | 0.29–8.07 | 0.61 | 0.97 | 0.28–3.41 | 0.97 | NA | NA | 1.00 |
| MTHFR | c.1298 A>C | rs1801131 | A/C | 0.32 | 1.17 | 0.21–6.52 | 0.86 | 0.36 | 0.07–1.73 | 0.20 | 3.37 | 0.55–20.70 | 0.19 |
| TYMS | 28bp repeats | rs34743033 | 2R–4R | 2R: 0.46 4R: 0.003 | 0.43 | 0.09–2.13 | 0.30 | 0.75 | 0.29–1.92 | 0.55 | 1.57 | 0.34–7.32 | 0.56 |
| TYMS | 6bp indel | rs34489327 | Ins/Del | 0.32 | 2.77 | 0.60–12.73 | 0.19 | 4.17 | 1.25–13.91 | 0.020 | NA | NA | 1.00 |
| DHFR hap1 | promoter region (−680–−317) | rs442767–rs1643641– rs1650695–rs1650696– rs408626 | A-A-T-A-G | 0.33 | 0.37 | 0.09–1.60 | 0.18 | 1.02 | 0.44–2.37 | 0.96 | 5.83 | 0.65–52.61 | 0.12 |
| DHFR hap2 | C-A-T-A-A | 0.30 | 8.07 | 0.97–67.5 | 0.054 | 1.42 | 0.62–3.26 | 0.41 | 1.18 | 0.24–5.65 | 0.84 | ||
| DHFR hap3 | C-A-T-A-G | 0.07 | 0.73 | 0.08–6.88 | 0.78 | 0.65 | 0.16–2.61 | 0.55 | 3.08 | 0.50–18.90 | 0.22 | ||
| DHFR hap4 | C-G-C-T-A | 0.27 | 1.15 | 0.30–4.41 | 0.84 | 1.19 | 0.52–2.73 | 0.68 | 0.49 | 0.09–2.70 | 0.41 | ||
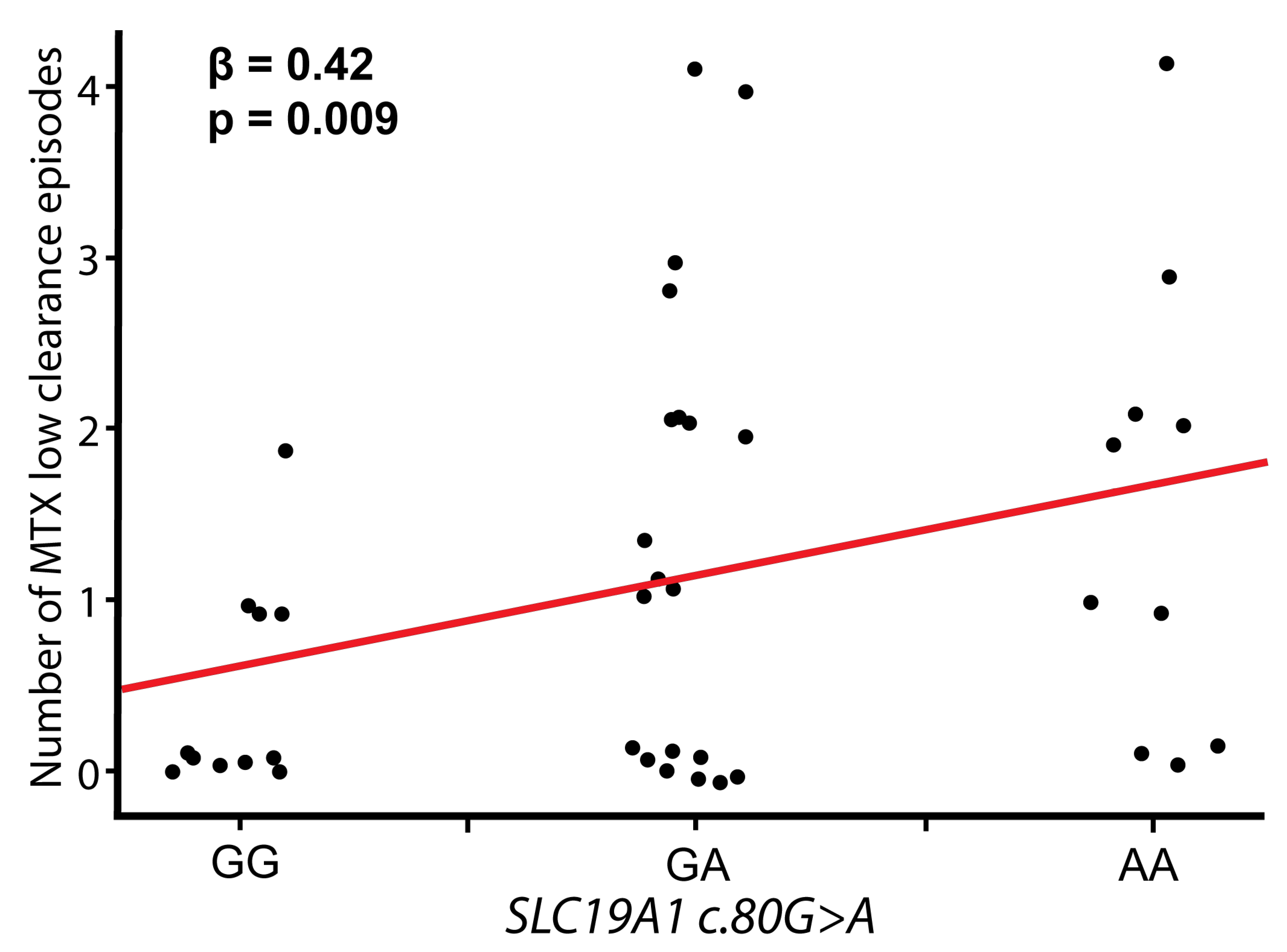
| SLC19A1 | c.80 G>A | ra1051266 | G/A | 0.50 | 1.09 | 0.26–4.57 | 0.90 | 1.38 | 0.56–3.40 | 0.49 | 9.70 | 1.70–55.78 | 0.011 |
| SLCO1B1 | c.521 T>C | rs4149056 | T/C | 0.16 | NA | NA | 1.00 | 1.65 | 0.27–10.02 | 0.59 | NA | NA | 1.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotur, N.; Lazic, J.; Ristivojevic, B.; Stankovic, B.; Gasic, V.; Dokmanovic, L.; Krstovski, N.; Milosevic, G.; Janic, D.; Zukic, B.; et al. Pharmacogenomic Markers of Methotrexate Response in the Consolidation Phase of Pediatric Acute Lymphoblastic Leukemia Treatment. Genes 2020, 11, 468. https://doi.org/10.3390/genes11040468
Kotur N, Lazic J, Ristivojevic B, Stankovic B, Gasic V, Dokmanovic L, Krstovski N, Milosevic G, Janic D, Zukic B, et al. Pharmacogenomic Markers of Methotrexate Response in the Consolidation Phase of Pediatric Acute Lymphoblastic Leukemia Treatment. Genes. 2020; 11(4):468. https://doi.org/10.3390/genes11040468
Chicago/Turabian StyleKotur, Nikola, Jelena Lazic, Bojan Ristivojevic, Biljana Stankovic, Vladimir Gasic, Lidija Dokmanovic, Nada Krstovski, Goran Milosevic, Dragana Janic, Branka Zukic, and et al. 2020. "Pharmacogenomic Markers of Methotrexate Response in the Consolidation Phase of Pediatric Acute Lymphoblastic Leukemia Treatment" Genes 11, no. 4: 468. https://doi.org/10.3390/genes11040468
APA StyleKotur, N., Lazic, J., Ristivojevic, B., Stankovic, B., Gasic, V., Dokmanovic, L., Krstovski, N., Milosevic, G., Janic, D., Zukic, B., & Pavlovic, S. (2020). Pharmacogenomic Markers of Methotrexate Response in the Consolidation Phase of Pediatric Acute Lymphoblastic Leukemia Treatment. Genes, 11(4), 468. https://doi.org/10.3390/genes11040468