A Study of Gene Expression Changes in Human Spinal and Oculomotor Neurons; Identifying Potential Links to Sporadic ALS


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
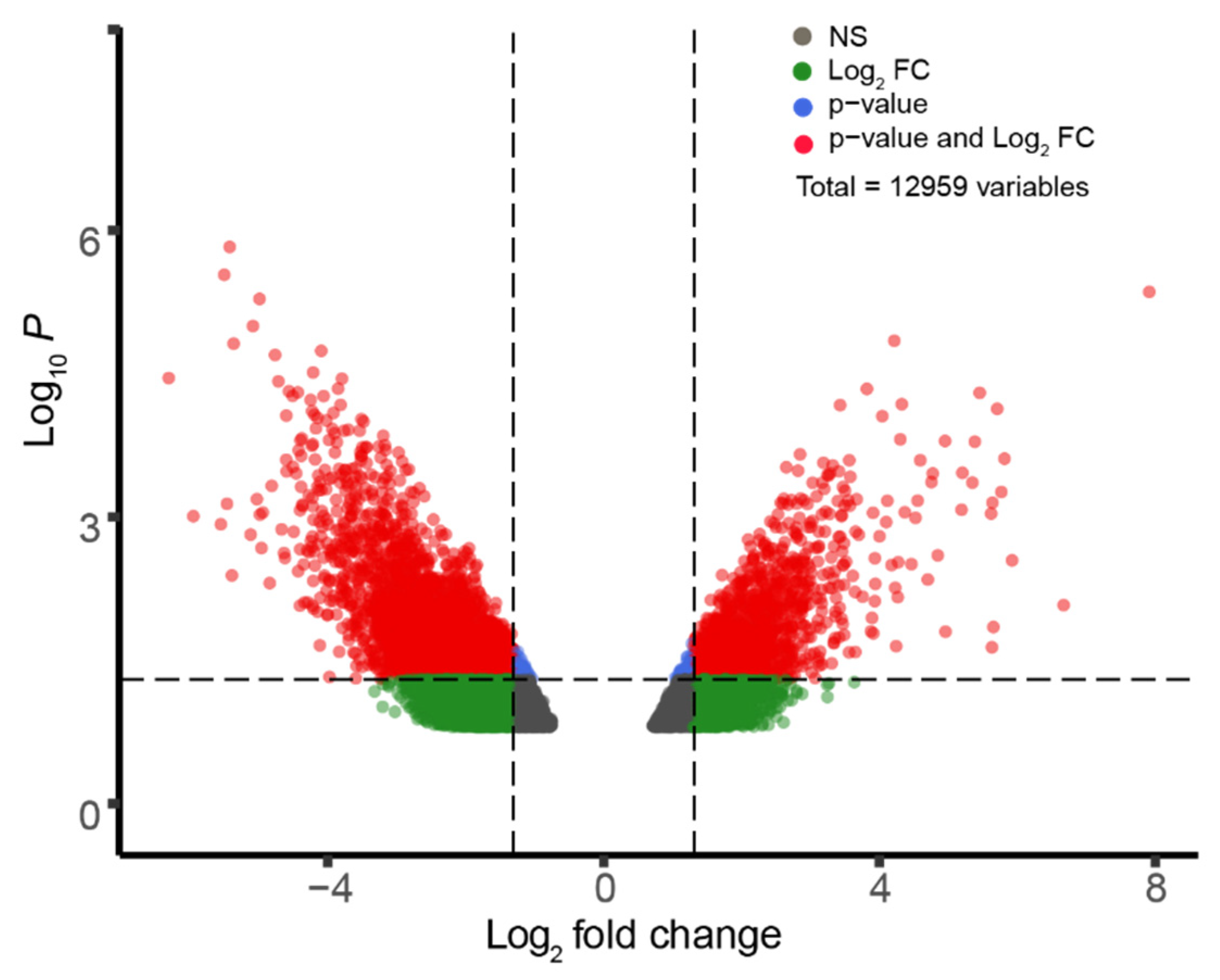
2.2. GEO2R Analysis and Volcano Plot Construction
2.3. Functional Enrichment Analysis with STRING and KEGG
3. Results
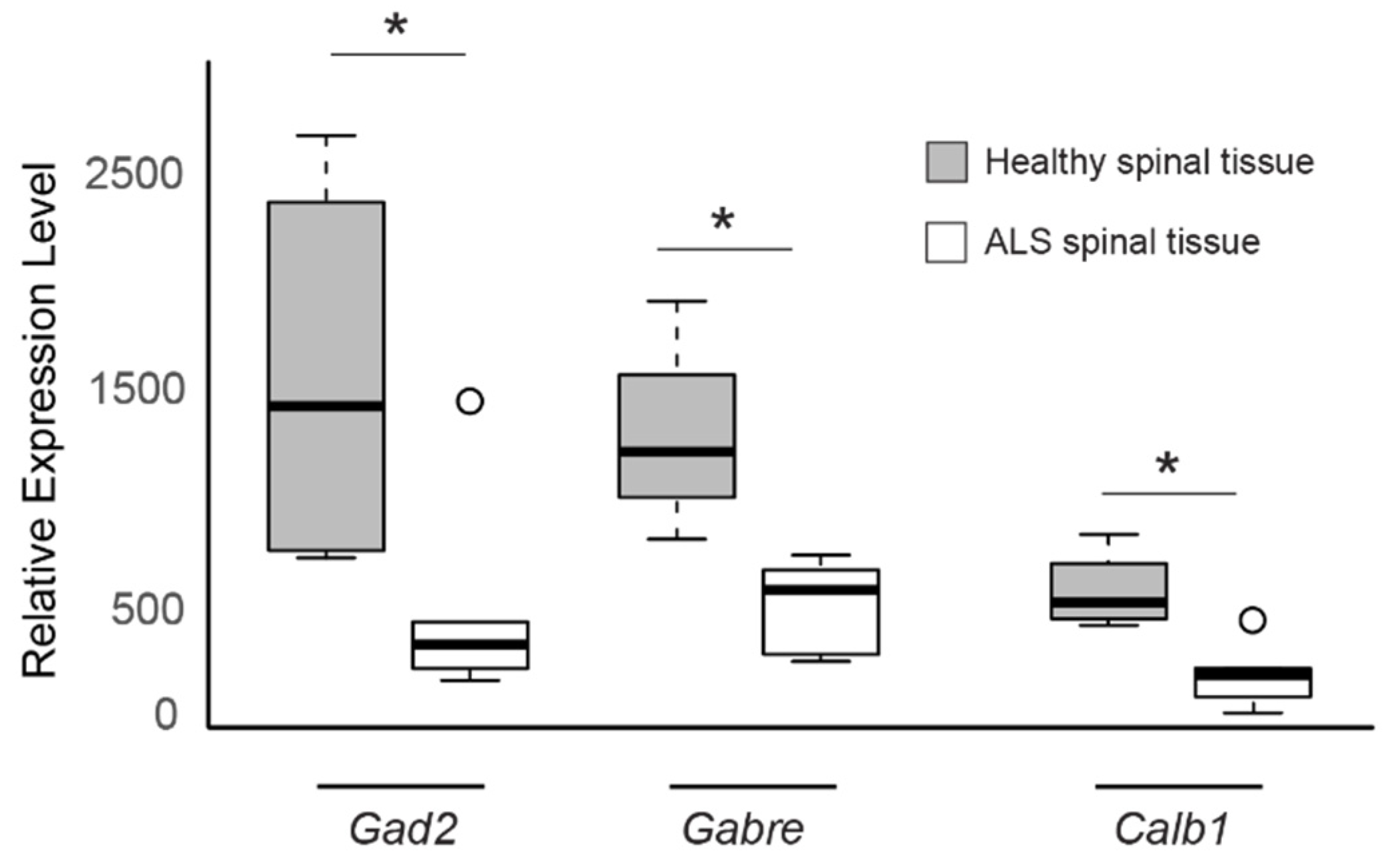
3.1. Differential Gene Expression in Sporadic ALS-Affected Spinal Tissue
3.2. Differential Gene Expression in Oculomotor Neurons of Healthy Patients
3.3. Identifying Common Genes across Both Datasets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Chio, A.; Borghero, G.; Restagno, G.; Mora, G.; Drepper, C.; Traynor, B.J.; Sendtner, M.; Brunetti, M.; Ossola, I.; Calvo, A.; et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 2012, 135, 784–793. [Google Scholar] [CrossRef]
- Andersen, P.M.; Nilsson, P.; Ala-Hurula, V.; Keranen, M.L.; Tarvainen, I.; Haltia, T.; Nilsson, L.; Binzer, M.; Forsgren, L.; Marklund, S.L. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat. Genet. 1995, 10, 61–66. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Foran, E.; Trotti, D. Glutamate Transporters and the Excitotoxic Path to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Antioxid. Redox Sign. 2009, 11, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal-Cord in Amyotrophic-Lateral-Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Aoki, M.; Pasinelli, P.; Berger, U.V.; Danbolt, N.C.; Brown, R.H.; Hediger, M.A. Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J. Biol. Chem. 2001, 276, 576–582. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Aizawa, H.; Tsuji, S.; Kakita, A.; Takahashi, H.; Kwak, S. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol. Dis. 2012, 45, 1121–1128. [Google Scholar] [CrossRef]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Hoyt, K.R.; Arden, S.R.; Aizenman, E.; Reynolds, I.J. Reverse Na+/Ca2+ exchange contributes to glutamate-induced intracellular Ca2+ concentration increases in cultured rat forebrain neurons. Mol. Pharmacol. 1998, 53, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Brittain, M.K.; Brustovetsky, T.; Sheets, P.L.; Brittain, J.M.; Khanna, R.; Cummins, T.R.; Brustovetsky, N. Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger. Neurobiol. Dis. 2012, 46, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.; Spiller, K.J.; Towne, C.; Kanning, K.C.; Choe, G.T.; Geber, A.; Akay, T.; Aebischer, P.; Henderson, C.E. Neuronal Matrix Metalloproteinase-9 is a Determinant of Selective Neurodegeneration. Neuron 2014, 81, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Taiana, M.; Rizzo, F.; Aguila Benitez, J.; Nijssen, J.; Allodi, I.; Melzi, V.; Bresolin, N.; Comi, G.P.; Hedlund, E.; et al. Synaptotagmin 13 is neuroprotective across motor neuron diseases. Acta Neuropathol. 2020. [Google Scholar] [CrossRef]
- Allodi, I.; Comley, L.; Nichterwitz, S.; Nizzardo, M.; Simone, C.; Benitez, J.A.; Cao, M.; Corti, S.; Hedlund, E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 2016, 6, 25960. [Google Scholar] [CrossRef]
- Liu, X.T.; Chen, J.H.; Guan, T.Y.; Yao, H.; Zhang, W.P.; Guan, Z.L.; Wang, Y.Q. miRNAs and target genes in the blood as biomarkers for the early diagnosis of Parkinson’s disease. BMC Syst. Biol. 2019, 13, 10. [Google Scholar] [CrossRef]
- Tang, H.X.; Zhang, Y.S. Identification and bioinformatics analysis of overlapping differentially expressed genes in depression, papillary thyroid cancer and uterine fibroids. Exp. Ther. Med. 2018, 15, 4810–4816. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Dangond, F.; Hwang, D.; Camelo, S.; Pasinelli, P.; Frosch, M.P.; Stephanopoulos, G.; Stephanopoulos, G.; Brown, R.H.; Gullans, S.R. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol. Genom. 2004, 16, 229–239. [Google Scholar] [CrossRef]
- Brockington, A.; Ning, K.; Heath, P.R.; Wood, E.; Kirby, J.; Fusi, N.; Lawrence, N.; Wharton, S.B.; Ince, P.G.; Shaw, P.J. Unravelling the enigma of selective vulnerability in neurodegeneration: Motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013, 125, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Daikhin, Y.; Horyn, O.; Nissim, I.; Nissim, I. Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia 2008, 49, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Holodkov, N.; Klima, R.; Grilli, F.; Guarnaccia, C.; Nizzardo, M.; Rizzo, F.; Garcia, R.; Feiguin, F. Downregulation of glutamic acid decarboxylase in Drosophila TDP-43-null brains provokes paralysis by affecting the organization of the neuromuscular synapses. Sci. Rep. 2018, 8, 1809. [Google Scholar] [CrossRef] [PubMed]
- Sigel, E.; Steinmann, M.E. Structure, Function, and Modulation of GABA(A) Receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef] [PubMed]
- Neelands, T.R.; Fisher, J.L.; Bianchi, M.; Macdonald, R.L. Spontaneous and gamma-aminobutyric acid (GABA)-activated GABA(A) receptor channels formed by epsilon subunit-containing isoforms. Mol. Pharmacol. 1999, 55, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Wafford, K.A.; Wingrove, P.; Ebert, B. Pharmacology of GABA(A) receptors exhibiting different levels of spontaneous activity. Eur. J. Pharmacol. 2003, 476, 17–24. [Google Scholar] [CrossRef]
- Schmidt, H. Three functional facets of calbindin D-28k. Front. Mol. Neurosci. 2012, 5, 25. [Google Scholar] [CrossRef]
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174. [Google Scholar] [CrossRef]
- Patai, R.; Nogradi, B.; Engelhardt, J.I.; Siklos, L. Calcium in the pathomechanism of amyotrophic lateral sclerosis—Taking center stage? Biochem. Biophys. Res. Commun. 2017, 483, 1031–1039. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Ho, B.K.; Mohamed, A.H.; Labella, V.; Smith, R.G.; Appel, S.H. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic-lateral-sclerosis. Ann. Neurol. 1994, 36, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Torres-Torrelo, J.; Rodriguez-Rosell, D.; Nunez-Abades, P.; Carrascal, L.; Torres, B. Glutamate modulates the firing rate in oculomotor nucleus motoneurons as a function of the recruitment threshold current. J. Physiol. Lond. 2012, 590, 3113–3127. [Google Scholar] [CrossRef] [PubMed]
- Carunchio, I.; Mollinari, C.; Pieri, M.; Merlo, D.; Zona, C. GABA(A) receptors present higher affinity and modified subunit composition in spinal motor neurons from a genetic model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2008, 28, 1275–1285. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, A.N.; Mathew, D. A Study of Gene Expression Changes in Human Spinal and Oculomotor Neurons; Identifying Potential Links to Sporadic ALS. Genes 2020, 11, 448. https://doi.org/10.3390/genes11040448
Patel AN, Mathew D. A Study of Gene Expression Changes in Human Spinal and Oculomotor Neurons; Identifying Potential Links to Sporadic ALS. Genes. 2020; 11(4):448. https://doi.org/10.3390/genes11040448
Chicago/Turabian StylePatel, Aayan N., and Dennis Mathew. 2020. "A Study of Gene Expression Changes in Human Spinal and Oculomotor Neurons; Identifying Potential Links to Sporadic ALS" Genes 11, no. 4: 448. https://doi.org/10.3390/genes11040448
APA StylePatel, A. N., & Mathew, D. (2020). A Study of Gene Expression Changes in Human Spinal and Oculomotor Neurons; Identifying Potential Links to Sporadic ALS. Genes, 11(4), 448. https://doi.org/10.3390/genes11040448