Transcriptomic Prediction of Pig Liver-Enriched Gene 1 Functions in a Liver Cell Line



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmid Preparation
2.3. RNA Extraction, Reverse-Transcription PCR (RT-PCR), Quantitative Real-Time PCR (qRT-PCR), and RNA-seq
2.4. Enrichment Analysis
2.5. Bioinformatic Prediction of the Functions of LEG1 Proteins
2.6. Statistics
2.7. Ethical Statement
3. Results
3.1. Bioinformatic Prediction of LEG1 Functions
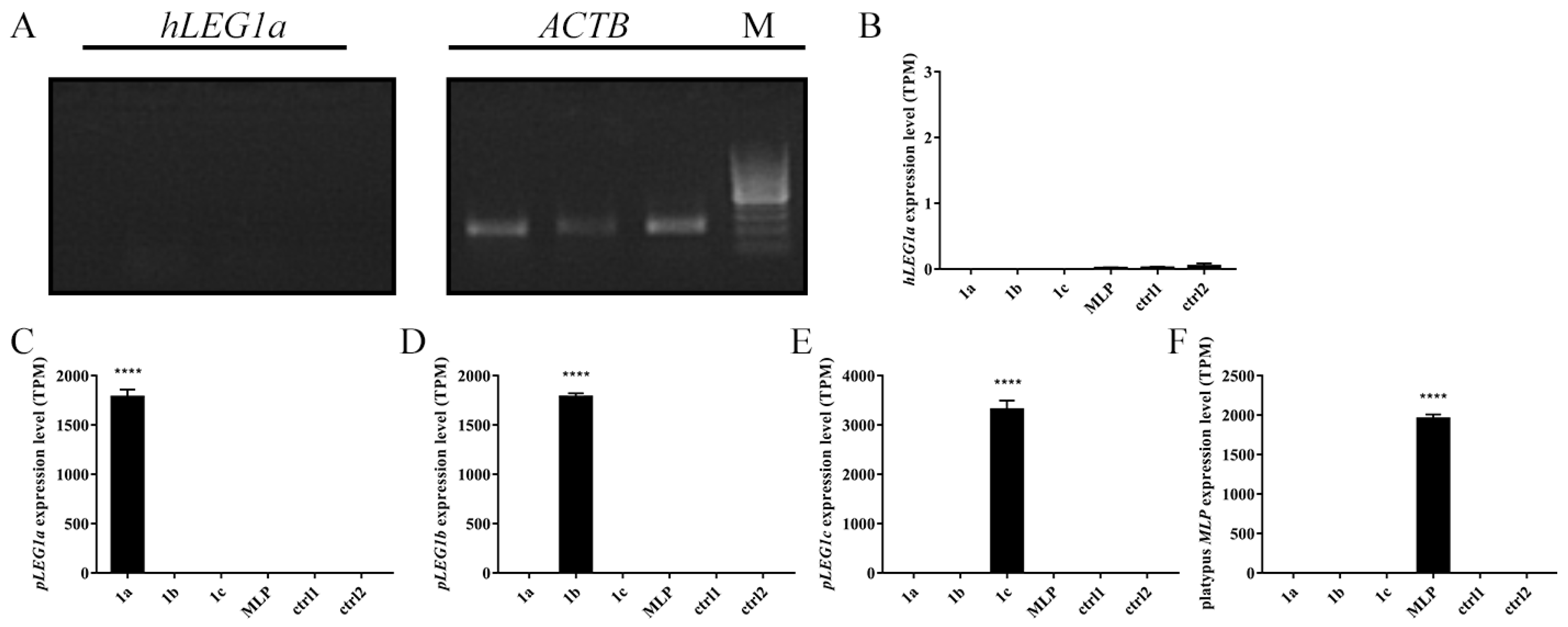
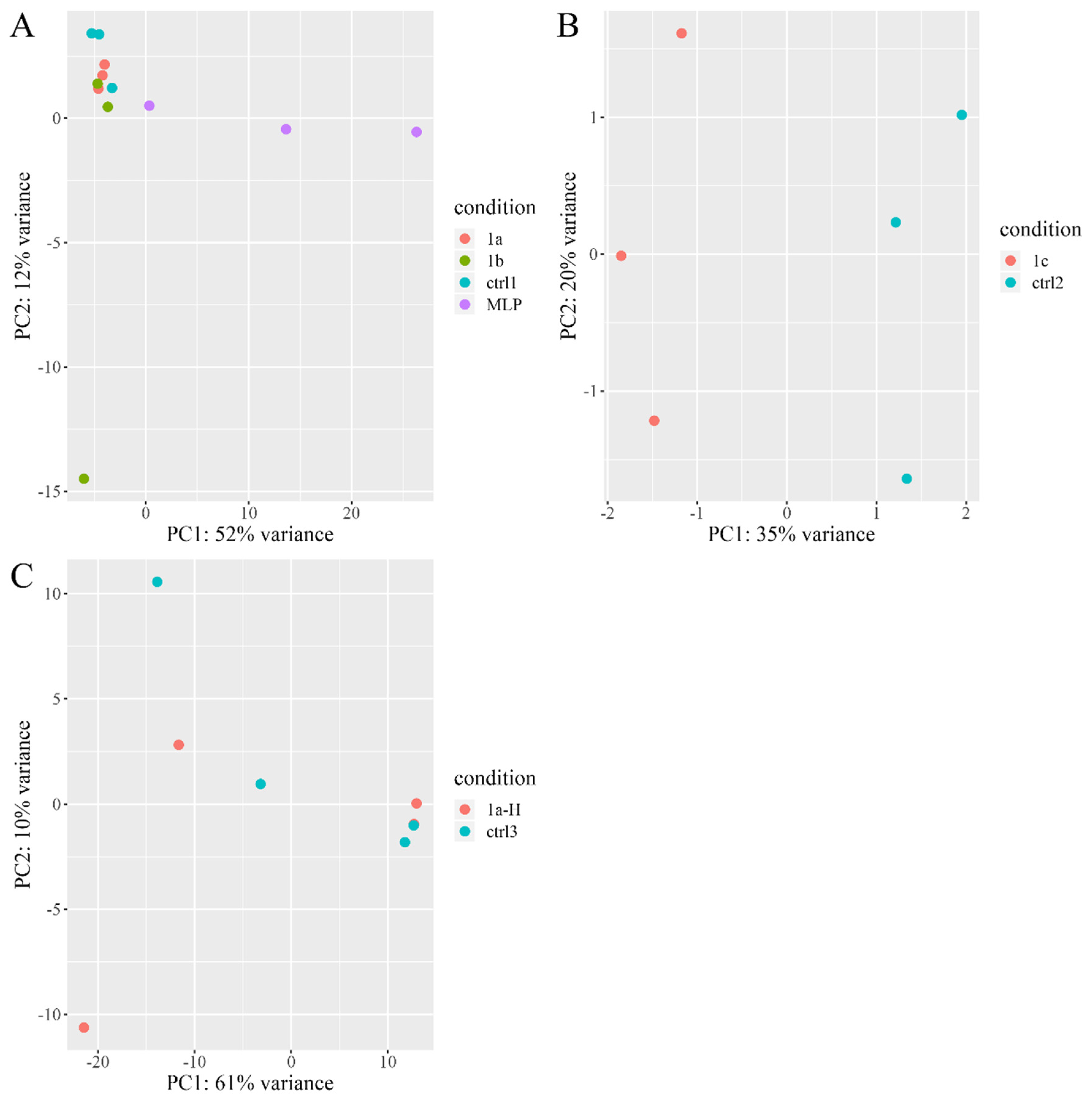
3.2. HepG2 Cells Transfected with LEG1 Genes
3.3. Enrichment Analysis Using DAVID and MSigD
3.4. GSEA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LEG1 | liver-enriched gene 1 |
| MLP | monotreme lactation protein |
| DUF781 | domain of unknown function 781 |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DEG | differentially expressed gene |
| BP | biological process |
| CC | cellular component |
| MF | molecular function |
| PCA | principal component analysis |
| DAVID | Database for Annotation Visualization and Integrated Discovery tool |
| MSigD | Molecular Signature Database |
| GSEA | Gene set enrichment analysis |
| TPM | transcripts per million |
| NES | Normalized enrichment score |
| ER | Endoplasmic reticulum |
References
- Chang, C.; Hu, M.; Zhu, Z.; Lo, L.J.; Chen, J.; Peng, J. Liver-enriched gene 1a and 1b Encode Novel Secretory Proteins Essential for Normal Liver Development in Zebrafish. PLOS ONE 2011, 6, e22910. [Google Scholar] [CrossRef]
- Hu, M.; Bai, Y.; Zhang, C.; Liu, F.; Cui, Z.; Chen, J.; Peng, J. Liver-Enriched Gene 1, a Glycosylated Secretory Protein, Binds to FGFR and Mediates an Anti-stress Pathway to Protect Liver Development in Zebrafish. PLoS Genet. 2016, 12, e1005881. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, B.; Deng, H.; Prinz, M.; Siegel, D. Body fluid identification by mass spectrometry. Int. J. Leg. Med. 2013, 127, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Boontheung, P.; Pang, E.; Yan, W.; Wong, D.T.; Loo, J.A. Comparison of N-linked Glycoproteins in Human Whole Saliva, Parotid, Submandibular, and Sublingual Glandular Secretions Identified using Hydrazide Chemistry and Mass Spectrometry. Clin. Proteom. 2008, 4, 80–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pini, T.; Leahy, T.; Soleilhavoup, C.; Tsikis, G.; Labas, V.; Combes-Soia, L.; Harichaux, G.; Rickard, J.; Druart, X.; De Graaf, S.P. Proteomic Investigation of Ram Spermatozoa and the Proteins Conferred by Seminal Plasma. J. Proteome Res. 2016, 15, 3700–3711. [Google Scholar] [CrossRef]
- Karn, R.C.; Chung, A.G.; Laukaitis, C.M. Shared and Unique Proteins in Human, Mouse and Rat Saliva Proteomes: Footprints of Functional Adaptation. Proteomes 2013, 1, 275–289. [Google Scholar] [CrossRef]
- Bisana, S.; Kumar, S.; Rismiller, P.; Nicol, S.C.; Lefevre, C.; Nicholas, K.R.; Sharp, J.A. Identification and Functional Characterization of a Novel Monotreme- Specific Antibacterial Protein Expressed during Lactation. PLOS ONE 2013, 8, e53686. [Google Scholar] [CrossRef]
- Enjapoori, A.K.; Grant, T.R.; Nicol, S.C.; Lefèvre, C.M.; Nicholas, K.R.; Sharp, J.A. Monotreme Lactation Protein Is Highly Expressed in Monotreme Milk and Provides Antimicrobial Protection. Genome Boil. Evol. 2014, 6, 2754–2773. [Google Scholar] [CrossRef]
- Dang, Y.; Wang, J.-Y.; Liu, C.; Zhang, K.; Jinrong, P.; He, J. Evolutionary and Molecular Characterization of liver-enriched gene 1. Sci. Rep. 2020, 10, 4262. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; A Irizarry, R.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 002832. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; A Blake, J.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, C.; Wang, Y.; Sun, Z.; Wang, N. PANDA: Protein function prediction using domain architecture and affinity propagation. Sci. Rep. 2018, 8, 3484. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Chou, C.-F.; Chung, H.-Y.; Chiang, C.-Y.; Li, C.-H.; Wu, J.-L.; Lin, H.-J.; Pai, T.-W.; Hu, C.-H.; Tzou, W.-S. Hypoxia-Inducible Factor 2 Alpha Is Essential for Hepatic Outgrowth and Functions via the Regulation of leg1 Transcription in the Zebrafish Embryo. PLOS ONE 2014, 9, e101980. [Google Scholar] [CrossRef]
- Zhu, Z.-H.; Hu, M.-J.; Chang, C.-Q.; Peng, J. Analysis of expression pattern of zebrafish leg1 homologus gene mu-leg1 in mouse. Hered. (Beijing) 2012, 34, 1174–1180. [Google Scholar]
- Freeman, T.C.; Ivens, A.; Baillie, J.K.; Beraldi, D.; Barnett, M.W.; Dorward, D.; Downing, A.; Fairbairn, L.; Kapetanovic, R.; Raza, S.; et al. A gene expression atlas of the domestic pig. BMC Boil. 2012, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Prince, V.; Pickett, F.B. Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet. 2002, 3, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of duplicate genes by complementary, degenerative mutations. Genet. 1999, 151, 1531–1545. [Google Scholar]
- Lin, L.-C.; Elkashty, O.; Ramamoorthi, M.; Trinh, N.; Liu, Y.; Sunavala-Dossabhoy, G.; Pranzatelli, T.; Michael, D.G.; Chivasso, C.; Perret, J.; et al. Cross-contamination of the human salivary gland HSG cell line with HeLa cells: A STR analysis study. Oral Dis. 2018, 24, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Phuchareon, J.; Ohta, Y.; Woo, J.M.; Eisele, D.W.; Tetsu, O. Genetic Profiling Reveals Cross-Contamination and Misidentification of 6 Adenoid Cystic Carcinoma Cell Lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLOS ONE 2009, 4, 6040. [Google Scholar] [CrossRef] [PubMed]
- Beucler, M.; Miller, W.E. Isolation of Salivary Epithelial Cells from Human Salivary Glands for In Vitro Growth as Salispheres or Monolayers. J. Vis. Exp. 2019, 149. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, R.; Liehr, T.; Kosyakova, N.; Rincic, M.; Azawi, S.S.H. Molecular cytogenetic characterization of two murine cancer cell lines derived from salivary gland. Boil. Commun. 2018, 63, 243–255. [Google Scholar] [CrossRef]
- Mehta, A.S.; Luz-Madrigal, A.; Li, J.-L.; Tsonis, P.A.; Singh, A. Comparative transcriptomic analysis and structure prediction of novel Newt proteins. PLOS ONE 2019, 14, e0220416. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Y.; Feng, X.; Luo, Z.; Kong, S.; Zhang, C.; Gong, A.; Yuan, H.; Cheng, L.; Wang, X. Genomic, molecular evolution, and expression analysis of NOX genes in soybean (Glycine max). Genom. 2019, 111, 619–628. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Y.; Liao, Z.; Xiong, J.; Wu, F.; Xu, J.; Lan, H.; Tang, Q.; Zhou, S.; Liu, Y.; et al. Evolutionary conservation and functional divergence of the LFK gene family play important roles in the photoperiodic flowering pathway of land plants. Hered. 2017, 120, 310–328. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Group | Category | Term/Pathway | p-Value |
|---|---|---|---|
| 1b | GO_BP | Negative regulation of transcription from RNA polymerase II promoter | 0.01 |
| GO_BP | Regulation of transcription from RNA polymerase II promoter | 0.03 | |
| GO_MF | Transcription factor activity, sequence-specific DNA binding | 0.027 | |
| 1c | GO_BP | Regulation of protein complex stability | 0.009 |
| GO_BP | autophagy | 0.018 | |
| GO_BP | Cell differentiation | 0.034 | |
| GO_BP | Positive regulation of glucose import | 0.045 | |
| GO_BP | Protein secretion | 0.048 | |
| GO_CC | Endoplasmic reticulum | 0.001 | |
| GO_CC | Endoplasmic reticulum chaperone complex | 0.017 | |
| GO_CC | Endoplasmic reticulum lumen | 0.035 | |
| GO_CC | Smooth endoplasmic reticulum | 0.039 | |
| GO_MF | Protein binding | 9 × 10−4 | |
| GO_MF | Unfolded protein binding | 0.01 | |
| GO_MF | Growth factor activity | 0.02 | |
| KEGG | Protein processing in endoplasmic reticulum | 2.9 × 10−4 | |
| MLP | GO_BP | translation | 1.6 × 10−13 |
| GO_BP | Mitochondrial translational termination | 4.2 × 10−6 | |
| GO_BP | Protein import into mitochondrial inner membrane | 0.002 | |
| GO_BP | Mitochondrial respiratory chain complex I assembly | 0.005 | |
| GO_BP | Cell cycle arrest | 0.014 | |
| GO_BP | Cerebral cortex development | 0.021 | |
| KEGG | Ribosome | 3.3 × 10−15 | |
| KEGG | Huntington’s disease | 2.6 × 10−4 | |
| KEGG | Alzheimer’s disease | 0.001 |
| Group | Up/Down Regulated DEGs | Category | Term/Pathway |
|---|---|---|---|
| 1a | Up | GO | Negative regulation of translational initiation |
| 1c | Up | GO | Regulation of response to endoplasmic reticulum stress |
| GO | Chaperone mediated protein complex assembly | ||
| GO | Smooth endoplasmic reticulum | ||
| GO | Cellular response to topologically incorrect protein | ||
| GO | Protein localization to synapse | ||
| GO | Protein folding in the endoplasmic reticulum | ||
| GO | Unfolded protein binding | ||
| GO | IRE1 mediated unfolded protein response | ||
| KEGG | Steroid hormone biosynthesis | ||
| Down | KEGG | Systemic lupus erythematosus | |
| MLP | Up | GO | Detoxification |
| GO | Defense response to fungus | ||
| GO | Defense response to Gram-negative bacterium | ||
| GO | Antigen processing and presentation | ||
| GO | Virion assembly | ||
| Down | GO | Low-density lipoprotein particle binding | |
| KEGG | Adherens junction |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Wang, Z.; Dang, Y.; Wang, J.; Jayaprakash, S.; Wang, H.; He, J. Transcriptomic Prediction of Pig Liver-Enriched Gene 1 Functions in a Liver Cell Line. Genes 2020, 11, 412. https://doi.org/10.3390/genes11040412
Zhang Z, Wang Z, Dang Y, Wang J, Jayaprakash S, Wang H, He J. Transcriptomic Prediction of Pig Liver-Enriched Gene 1 Functions in a Liver Cell Line. Genes. 2020; 11(4):412. https://doi.org/10.3390/genes11040412
Chicago/Turabian StyleZhang, Zhe, Zizengchen Wang, Yanna Dang, Jinyang Wang, Sakthidasan Jayaprakash, Huanan Wang, and Jin He. 2020. "Transcriptomic Prediction of Pig Liver-Enriched Gene 1 Functions in a Liver Cell Line" Genes 11, no. 4: 412. https://doi.org/10.3390/genes11040412
APA StyleZhang, Z., Wang, Z., Dang, Y., Wang, J., Jayaprakash, S., Wang, H., & He, J. (2020). Transcriptomic Prediction of Pig Liver-Enriched Gene 1 Functions in a Liver Cell Line. Genes, 11(4), 412. https://doi.org/10.3390/genes11040412