TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease

Abstract
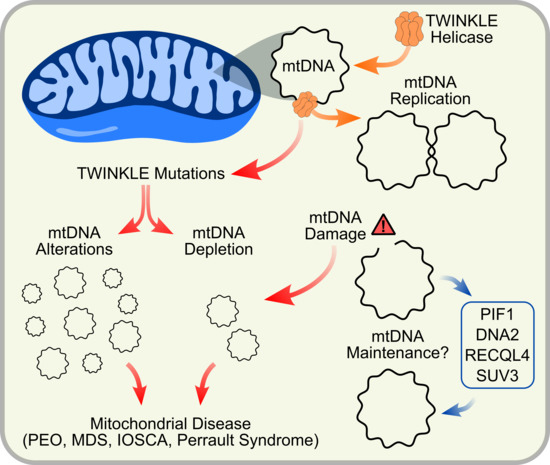
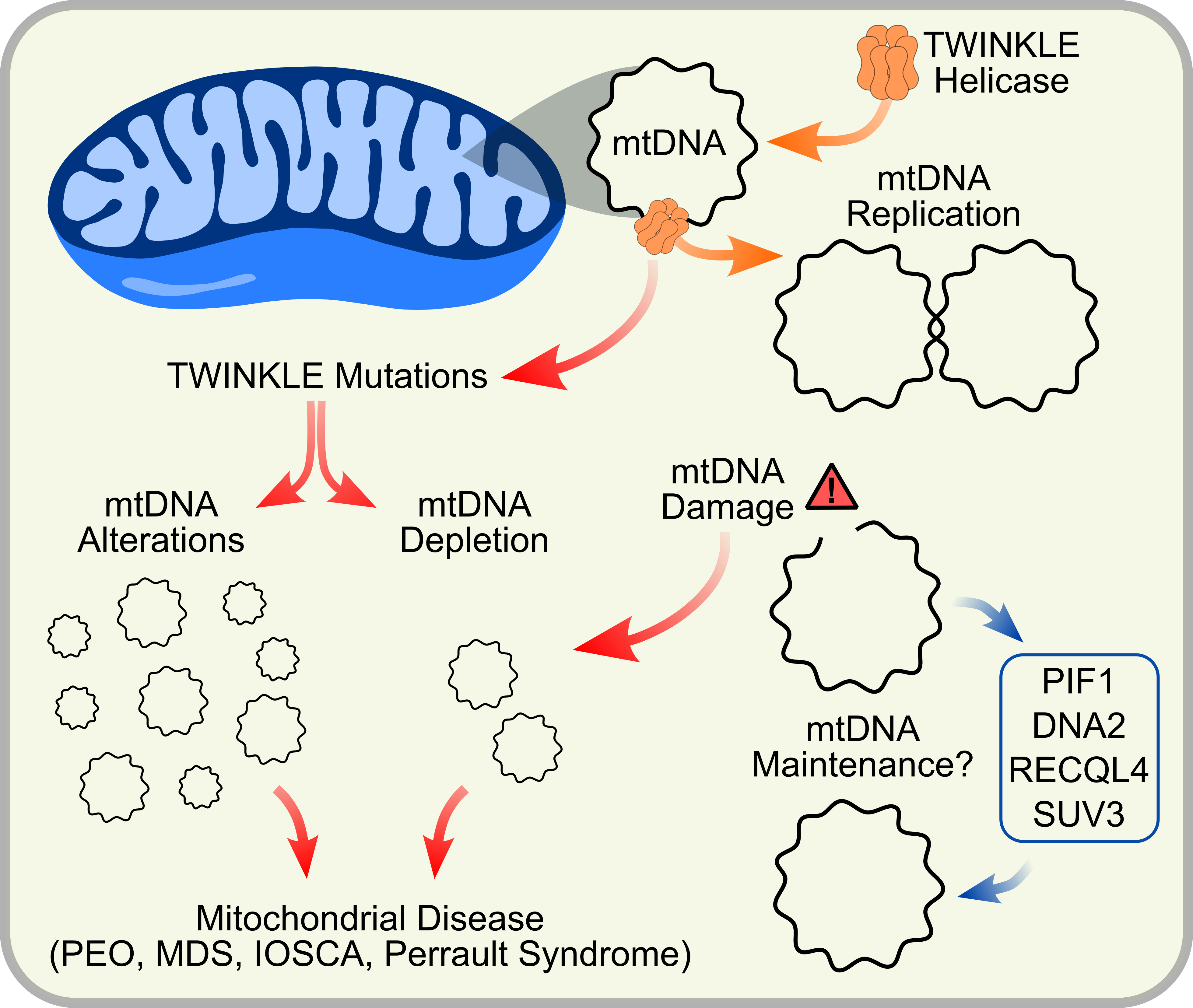{kind=link}
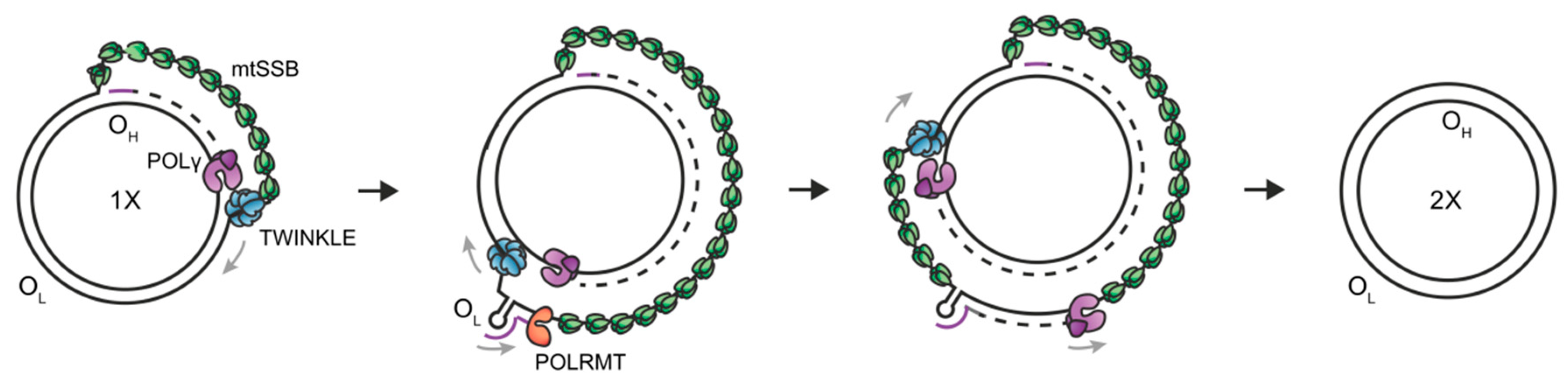{kind=link}
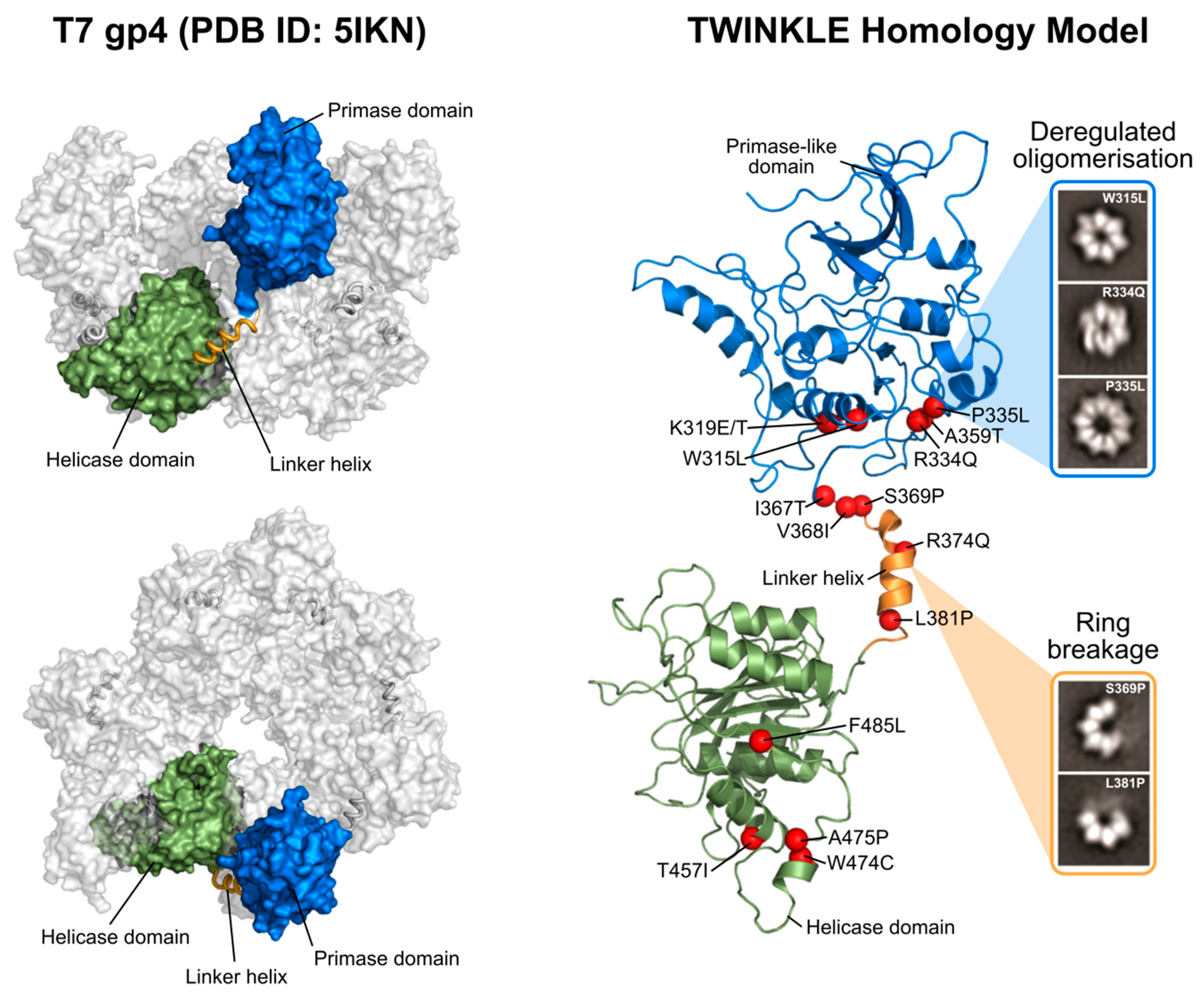{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Overview of mtDNA Replication
2. TWINKLE Helicase
2.1. Structure and Helicase Activity
2.1.1. Structure and Domain Organization
2.1.2. TWINKLE as the Sole Replicative Helicase
2.1.3. Additional Proposed Functions of TWINKLE
2.1.4. TWINKLE in the Context of the Replisome
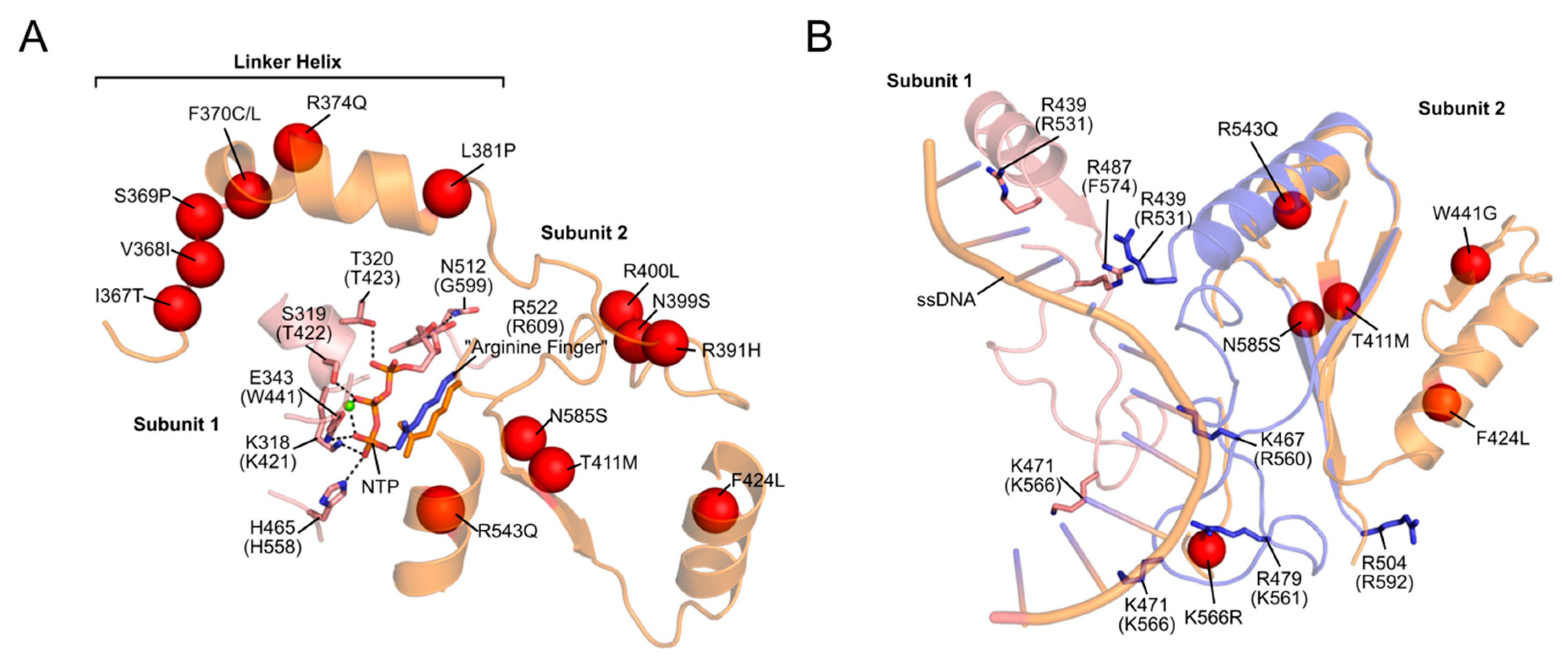
2.2. Disease
2.2.1. Progressive External Ophthalmoplegia (PEO)
2.2.2. Other Diseases
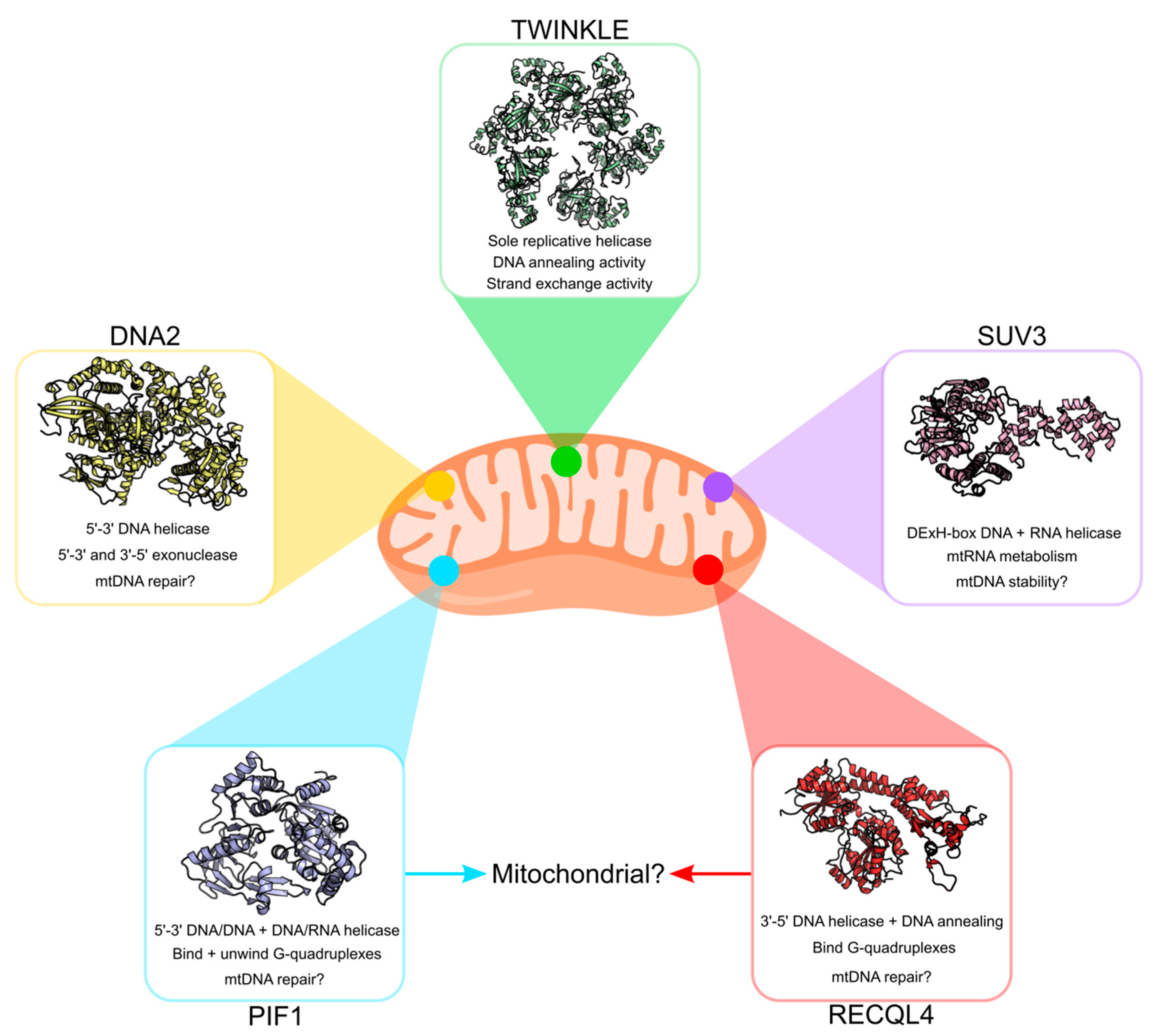
3. Other Potential Mitochondrial DNA Helicases
3.1. PIF1 Helicase
3.2. DNA2 Nuclease/Helicase
3.3. RECQL4 Helicase
3.4. SUV3 and Other RNA Helicases
4. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gray, H.; Wong, T.W. Purification and identification of subunit structure of the human mitochondrial DNA polymerase. J. Biol. Chem. 1992, 267, 5835–5841. [Google Scholar] [PubMed]
- Fan, L.; Kim, S.; Farr, C.L.; Schaefer, K.T.; Randolph, K.M.; Tainer, J.A.; Kaguni, L.S. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J. Mol. Biol. 2006, 358, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE Has 5’-> 3’ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef]
- Farr, C.L.; Wang, Y.; Kaguni, L.S. Functional interactions of mitochondrial DNA polymerase and single-stranded DNA-binding protein. Template-primer DNA binding and initiation and elongation of DNA strand synthesis. J. Biol. Chem. 1999, 274, 14779–14785. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef]
- Tsurumi, C.; Yoshihara, Y.; Osaka, F.; Yamada, F.; Tani, I.; Higuti, T.; Shimizu, M.; Oeda, K.; Ohkawa, H.; Toda, H.; et al. cDNA cloning and sequencing for the import precursor of subunit B in H(+)-ATP synthase from rat mitochondria. Biochem. Biophys. Res. Commun. 1990, 169, 136–142. [Google Scholar] [CrossRef]
- Wanrooij, S.; Fuste, J.M.; Farge, G.; Shi, Y.; Gustafsson, C.M.; Falkenberg, M. Human mitochondrial RNA polymerase primes lagging-strand DNA synthesis in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 11122–11127. [Google Scholar] [CrossRef]
- Robberson, D.L.; Clayton, D.A. Replication of mitochondrial DNA in mouse L cells and their thymidine kinase - derivatives: Displacement replication on a covalently-closed circular template. Proc. Natl. Acad. Sci. USA 1972, 69, 3810–3814. [Google Scholar] [CrossRef]
- Tapper, D.P.; Clayton, D.A. Mechanism of replication of human mitochondrial DNA. Localization of the 5’ ends of nascent daughter strands. J. Biol. Chem. 1981, 256, 5109–5115. [Google Scholar]
- Fuste, J.M.; Wanrooij, S.; Jemt, E.; Granycome, C.E.; Cluett, T.J.; Shi, Y.; Atanassova, N.; Holt, I.J.; Gustafsson, C.M.; Falkenberg, M. Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol. Cell 2010, 37, 67–78. [Google Scholar] [CrossRef]
- Miralles Fuste, J.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, O.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef]
- Pyle, A.M. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 2008, 37, 317–336. [Google Scholar] [CrossRef]
- Lohman, T.M.; Tomko, E.J.; Wu, C.G. Non-hexameric DNA helicases and translocases: Mechanisms and regulation. Nat. Rev. Mol. Cell Biol. 2008, 9, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef] [PubMed]
- Donmez, I.; Patel, S.S. Mechanisms of a ring shaped helicase. Nucleic Acids Res. 2006, 34, 4216–4224. [Google Scholar] [CrossRef] [PubMed]
- Fairman-Williams, M.E.; Guenther, U.P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef]
- Singleton, M.R.; Sawaya, M.R.; Ellenberger, T.; Wigley, D.B. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 2000, 101, 589–600. [Google Scholar] [CrossRef]
- Picha, K.M.; Ahnert, P.; Patel, S.S. DNA binding in the central channel of bacteriophage T7 helicase-primase is a multistep process. Nucleotide hydrolysis is not required. Biochemistry 2000, 39, 6401–6409. [Google Scholar] [CrossRef]
- Crampton, D.J.; Ohi, M.; Qimron, U.; Walz, T.; Richardson, C.C. Oligomeric states of bacteriophage T7 gene 4 primase/helicase. J. Mol. Biol. 2006, 360, 667–677. [Google Scholar] [CrossRef]
- Bailey, S.; Eliason, W.K.; Steitz, T.A. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 2007, 318, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Richardson, C.C. Single-stranded DNA binding protein and DNA helicase of bacteriophage T7 mediate homologous DNA strand exchange. EMBO J. 1996, 15, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Marintcheva, B.; Hamdan, S.M.; Richardson, C.C. The C-terminal residues of bacteriophage T7 gene 4 helicase-primase coordinate helicase and DNA polymerase activities. J. Biol. Chem. 2006, 281, 25841–25849. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.M.; Johnson, D.E.; Tanner, N.A.; Lee, J.B.; Qimron, U.; Tabor, S.; van Oijen, A.M.; Richardson, C.C. Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol. Cell 2007, 27, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.E.; Gray, M.W. Bacteriophage origins of mitochondrial replication and transcription proteins. Trends Genet. 2006, 22, 90–95. [Google Scholar] [CrossRef]
- Lee, S.J.; Zhu, B.; Akabayov, B.; Richardson, C.C. Zinc-binding domain of the bacteriophage T7 DNA primase modulates binding to the DNA template. J. Biol. Chem. 2012, 287, 39030–39040. [Google Scholar] [CrossRef]
- Ziebarth, T.D.; Farr, C.L.; Kaguni, L.S. Modular architecture of the hexameric human mitochondrial DNA helicase. J. Mol. Biol. 2007, 367, 1382–1391. [Google Scholar] [CrossRef]
- Holmlund, T.; Farge, G.; Pande, V.; Korhonen, J.; Nilsson, L.; Falkenberg, M. Structure-function defects of the twinkle amino-terminal region in progressive external ophthalmoplegia. Biochim. Biophys. Acta 2009, 1792, 132–139. [Google Scholar] [CrossRef]
- Kusakabe, T.; Richardson, C.C. The role of the zinc motif in sequence recognition by DNA primases. J. Biol. Chem. 1996, 271, 19563–19570. [Google Scholar] [CrossRef]
- Matsushima, Y.; Kaguni, L.S. Differential phenotypes of active site and human autosomal dominant progressive external ophthalmoplegia mutations in Drosophila mitochondrial DNA helicase expressed in Schneider cells. J. Biol. Chem. 2007, 282, 9436–9444. [Google Scholar] [CrossRef]
- Farge, G.; Holmlund, T.; Khvorostova, J.; Rofougaran, R.; Hofer, A.; Falkenberg, M. The N-terminal domain of TWINKLE contributes to single-stranded DNA binding and DNA helicase activities. Nucleic Acids Res. 2008, 36, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Guo, S.; Tabor, S.; Richardson, C.C.; Ellenberger, T. Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell 1999, 99, 167–177. [Google Scholar] [CrossRef]
- Toth, E.A.; Li, Y.; Sawaya, M.R.; Cheng, Y.; Ellenberger, T. The crystal structure of the bifunctional primase-helicase of bacteriophage T7. Mol. Cell 2003, 12, 1113–1123. [Google Scholar] [CrossRef]
- Fernandez-Millan, P.; Lazaro, M.; Cansiz-Arda, S.; Gerhold, J.M.; Rajala, N.; Schmitz, C.A.; Silva-Espina, C.; Gil, D.; Bernado, P.; Valle, M.; et al. The hexameric structure of the human mitochondrial replicative helicase Twinkle. Nucleic Acids Res. 2015, 43, 4284–4295. [Google Scholar] [CrossRef]
- Peter, B.; Farge, G.; Pardo-Hernandez, C.; Tangefjord, S.; Falkenberg, M. Structural basis for adPEO-causing mutations in the mitochondrial TWINKLE helicase. Hum. Mol. Genet. 2019, 28, 1090–1099. [Google Scholar] [CrossRef]
- Crampton, D.J.; Guo, S.; Johnson, D.E.; Richardson, C.C. The arginine finger of bacteriophage T7 gene 4 helicase: Role in energy coupling. Proc. Natl. Acad. Sci. USA 2004, 101, 4373–4378. [Google Scholar] [CrossRef]
- Guo, S.; Tabor, S.; Richardson, C.C. The linker region between the helicase and primase domains of the bacteriophage T7 gene 4 protein is critical for hexamer formation. J. Biol. Chem. 1999, 274, 30303–30309. [Google Scholar] [CrossRef]
- Lee, S.J.; Richardson, C.C. The linker region between the helicase and primase domains of the gene 4 protein of bacteriophage T7. Role in helicase conformation and activity. J. Biol. Chem. 2004, 279, 23384–23393. [Google Scholar] [CrossRef]
- Korhonen, J.A.; Pande, V.; Holmlund, T.; Farge, G.; Pham, X.H.; Nilsson, L.; Falkenberg, M. Structure-function defects of the TWINKLE linker region in progressive external ophthalmoplegia. J. Mol. Biol. 2008, 377, 691–705. [Google Scholar] [CrossRef]
- Goffart, S.; Cooper, H.M.; Tyynismaa, H.; Wanrooij, S.; Suomalainen, A.; Spelbrink, J.N. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009, 18, 328–340. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Chanson, J.B.; Wilhelm, J.M.; Sellal, F.; Mayencon, M.; Mohr, M.; Tranchant, C.; Mousson de Camaret, B. A novel variation in the Twinkle linker region causing late-onset dementia. Neurogenetics 2010, 11, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Ziebarth, T.D.; Gonzalez-Soltero, R.; Makowska-Grzyska, M.M.; Nunez-Ramirez, R.; Carazo, J.M.; Kaguni, L.S. Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J. Biol. Chem. 2010, 285, 14639–14647. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Matic, S.; Kuhl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef] [PubMed]
- Jemt, E.; Farge, G.; Backstrom, S.; Holmlund, T.; Gustafsson, C.M.; Falkenberg, M. The mitochondrial DNA helicase TWINKLE can assemble on a closed circular template and support initiation of DNA synthesis. Nucleic Acids Res. 2011, 39, 9238–9249. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Picha, K.M. Structure and function of hexameric helicases. Annu. Rev. Biochem. 2000, 69, 651–697. [Google Scholar] [CrossRef]
- O’Shea, V.L.; Berger, J.M. Loading strategies of ring-shaped nucleic acid translocases and helicases. Curr. Opin. Struct. Biol. 2014, 25, 16–24. [Google Scholar] [CrossRef]
- Kaur, P.; Longley, M.J.; Pan, H.; Wang, W.; Countryman, P.; Wang, H.; Copeland, W.C. Single-molecule level structural dynamics of DNA unwinding by human mitochondrial Twinkle helicase. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Ahnert, P.; Patel, S.S. Asymmetric interactions of hexameric bacteriophage T7 DNA helicase with the 5’- and 3’-tails of the forked DNA substrate. J. Biol. Chem. 1997, 272, 32267–32273. [Google Scholar] [CrossRef]
- Sen, D.; Nandakumar, D.; Tang, G.Q.; Patel, S.S. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J. Biol. Chem. 2012, 287, 14545–14556. [Google Scholar] [CrossRef]
- Sen, D.; Patel, G.; Patel, S.S. Homologous DNA strand exchange activity of the human mitochondrial DNA helicase TWINKLE. Nucleic Acids Res. 2016, 44, 4200–4210. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Crouch, J.D.; Bharti, S.K.; Sommers, J.A.; Carney, S.M.; Yakubovskaya, E.; Garcia-Diaz, M.; Trakselis, M.A.; Brosh, R.M., Jr. Biochemical Characterization of the Human Mitochondrial Replicative Twinkle Helicase: SUBSTRATE SPECIFICITY, DNA BRANCH MIGRATION, AND ABILITY TO OVERCOME BLOCKADES TO DNA UNWINDING. J. Biol. Chem. 2016, 291, 14324–14339. [Google Scholar] [CrossRef] [PubMed]
- Weissman, L.; de Souza-Pinto, N.C.; Stevnsner, T.; Bohr, V.A. DNA repair, mitochondria, and neurodegeneration. Neuroscience 2007, 145, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, M.; Guo, Z.; Lu, H.; Qian, L.; Dai, H.; Qiu, J.; Yakubovskaya, E.; Bogenhagen, D.F.; Demple, B.; et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell 2008, 32, 325–336. [Google Scholar] [CrossRef]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Howell, N. Mutational analysis of the human mitochondrial genome branches into the realm of bacterial genetics. Am. J. Hum. Genet. 1996, 59, 749–755. [Google Scholar]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Mori, M.P.; de Souza-Pinto, N.C. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 2014, 17, 164–181. [Google Scholar] [CrossRef]
- Linnane, A.W.; Marzuki, S.; Ozawa, T.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 1, 642–645. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef]
- Chen, X.J. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol Mol Biol Rev 2013, 77, 476–496. [Google Scholar] [CrossRef]
- Hagstrom, E.; Freyer, C.; Battersby, B.J.; Stewart, J.B.; Larsson, N.G. No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res. 2014, 42, 1111–1116. [Google Scholar] [CrossRef]
- Bogenhagen, D.; Clayton, D.A. Mechanism of mitochondrial DNA replication in mouse L-cells: Introduction of superhelical turns into newly replicated molecules. J. Mol. Biol. 1978, 119, 69–81. [Google Scholar] [CrossRef]
- Doda, J.N.; Wright, C.T.; Clayton, D.A. Elongation of displacement-loop strands in human and mouse mitochondrial DNA is arrested near specific template sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 6116–6120. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef]
- Ylikallio, E.; Tyynismaa, H.; Tsutsui, H.; Ide, T.; Suomalainen, A. High mitochondrial DNA copy number has detrimental effects in mice. Hum. Mol. Genet. 2010, 19, 2695–2705. [Google Scholar] [CrossRef]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N. Functional organization of mammalian mitochondrial DNA in nucleoids: History, recent developments, and future challenges. IUBMB Life 2010, 62, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed]
- Kulczyk, A.W.; Moeller, A.; Meyer, P.; Sliz, P.; Richardson, C.C. Cryo-EM structure of the replisome reveals multiple interactions coordinating DNA synthesis. Proc. Natl. Acad. Sci. USA 2017, 114, E1848–E1856. [Google Scholar] [CrossRef]
- Wallen, J.R.; Zhang, H.; Weis, C.; Cui, W.; Foster, B.M.; Ho, C.M.W.; Hammel, M.; Tainer, J.A.; Gross, M.L.; Ellenberger, T. Hybrid Methods Reveal Multiple Flexibly Linked DNA Polymerases within the Bacteriophage T7 Replisome. Structure 2017, 25, 157–166. [Google Scholar] [CrossRef]
- Gao, Y.; Cui, Y.; Fox, T.; Lin, S.; Wang, H.; de Val, N.; Zhou, Z.H.; Yang, W. Structures and operating principles of the replisome. Science 2019, 363. [Google Scholar] [CrossRef]
- Patel, S.S.; Pandey, M.; Nandakumar, D. Dynamic coupling between the motors of DNA replication: Hexameric helicase, DNA polymerase, and primase. Curr. Opin. Chem. Biol. 2011, 15, 595–605. [Google Scholar] [CrossRef]
- Notarnicola, S.M.; Mulcahy, H.L.; Lee, J.; Richardson, C.C. The acidic carboxyl terminus of the bacteriophage T7 gene 4 helicase/primase interacts with T7 DNA polymerase. J. Biol. Chem. 1997, 272, 18425–18433. [Google Scholar] [CrossRef]
- Delagoutte, E.; von Hippel, P.H. Molecular mechanisms of the functional coupling of the helicase (gp41) and polymerase (gp43) of bacteriophage T4 within the DNA replication fork. Biochemistry 2001, 40, 4459–4477. [Google Scholar] [CrossRef]
- Stano, N.M.; Jeong, Y.J.; Donmez, I.; Tummalapalli, P.; Levin, M.K.; Patel, S.S. DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature 2005, 435, 370–373. [Google Scholar] [CrossRef]
- Loparo, J.J.; Kulczyk, A.W.; Richardson, C.C.; van Oijen, A.M. Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc Natl Acad Sci U S A 2011, 108, 3584–3589. [Google Scholar] [CrossRef] [PubMed]
- Frick, D.N.; Richardson, C.C. DNA primases. Annu. Rev. Biochem. 2001, 70, 39–80. [Google Scholar] [CrossRef] [PubMed]
- Corn, J.E.; Pelton, J.G.; Berger, J.M. Identification of a DNA primase template tracking site redefines the geometry of primer synthesis. Nat. Struct. Mol. Biol. 2008, 15, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.T.; Kaguni, L.S. Reduced stimulation of recombinant DNA polymerase γ and mitochondrial DNA (mtDNA) helicase by variants of mitochondrial single-stranded DNA-binding protein (mtSSB) correlates with defects in mtDNA replication in animal cells. J. Biol. Chem. 2011, 286, 40649–40658. [Google Scholar] [CrossRef] [PubMed]
- Tanguy Le Gac, N.; Villani, G.; Hoffmann, J.S.; Boehmer, P.E. The UL8 subunit of the herpes simplex virus type-1 DNA helicase-primase optimizes utilization of DNA templates covered by the homologous single-strand DNA-binding protein ICP8. J. Biol. Chem. 1996, 271, 21645–21651. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Bushnell, D.A.; Elias, P.; Lehman, I.R. The UL8 subunit of the heterotrimeric herpes simplex virus type 1 helicase-primase is required for the unwinding of single strand DNA-binding protein (ICP8)-coated DNA substrates. J. Biol. Chem. 1997, 272, 22766–22770. [Google Scholar] [CrossRef]
- Ramanagoudr-Bhojappa, R.; Blair, L.P.; Tackett, A.J.; Raney, K.D. Physical and functional interaction between yeast Pif1 helicase and Rim1 single-stranded DNA binding protein. Nucleic Acids Res. 2013, 41, 1029–1046. [Google Scholar] [CrossRef]
- Del Dotto, V.; Ullah, F.; Di Meo, I.; Magini, P.; Gusic, M.; Maresca, A.; Caporali, L.; Palombo, F.; Tagliavini, F.; Baugh, E.H.; et al. SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J. Clin. Investig. 2019. [Google Scholar] [CrossRef]
- Piro-Mégy, C.; Sarzi, E.; Tarrés-Solé, A.; Péquignot, M.; Hensen, F.; Quilès, M.; Manes, G.; Chakraborty, A.; Sénéchal, A.; Bocquet, B.; et al. Dominant mutations in mtDNA maintenance gene SSBP1 cause optic atrophy and foveopathy. J. Clin. Investig. 2019. [Google Scholar] [CrossRef]
- Rajala, N.; Gerhold, J.M.; Martinsson, P.; Klymov, A.; Spelbrink, J.N. Replication factors transiently associate with mtDNA at the mitochondrial inner membrane to facilitate replication. Nucleic Acids Res. 2014, 42, 952–967. [Google Scholar] [CrossRef]
- Albring, M.; Griffith, J.; Attardi, G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl. Acad. Sci. USA 1977, 74, 1348–1352. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Silva Ramos, E.; Motori, E.; Brüser, C.; Kühl, I.; Yeroslaviz, A.; Ruzzenente, B.; Kauppila, J.H.K.; Busch, J.D.; Hultenby, K.; Habermann, B.H.; et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLOS Genet. 2019. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Young, M.J.; Copeland, W.C. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 2016, 38, 52–62. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA transcription: Regulating the power supply. Cell 2007, 130, 211–213. [Google Scholar] [CrossRef][Green Version]
- Fratter, C.; Gorman, G.S.; Stewart, J.D.; Buddles, M.; Smith, C.; Evans, J.; Seller, A.; Poulton, J.; Roberts, M.; Hanna, M.G.; et al. The clinical, histochemical, and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology 2010, 74, 1619–1626. [Google Scholar] [CrossRef]
- Hakonen, A.H.; Goffart, S.; Marjavaara, S.; Paetau, A.; Cooper, H.; Mattila, K.; Lampinen, M.; Sajantila, A.; Lonnqvist, T.; Spelbrink, J.N.; et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum. Mol. Genet. 2008, 17, 3822–3835. [Google Scholar] [CrossRef]
- Hudson, G.; Keers, S.; Yu-Wai-Man, P.; Griffiths, P.; Huoponen, K.; Savontaus, M.L.; Nikoskelainen, E.; Zeviani, M.; Carrara, F.; Horvath, R.; et al. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am. J. Hum. Genet. 2005, 77, 1086–1091. [Google Scholar] [CrossRef]
- Lonnqvist, T.; Paetau, A.; Valanne, L.; Pihko, H. Recessive twinkle mutations cause severe epileptic encephalopathy. Brain 2009, 132, 1553–1562. [Google Scholar] [CrossRef]
- Van Hove, J.L.; Cunningham, V.; Rice, C.; Ringel, S.P.; Zhang, Q.; Chou, P.C.; Truong, C.K.; Wong, L.J. Finding twinkle in the eyes of a 71-year-old lady: A case report and review of the genotypic and phenotypic spectrum of TWINKLE-related dominant disease. Am. J. Med. Genet. A 2009, 149A, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man. Available online: https://omim.org/ (accessed on 9 April 2020).
- McClelland, C.; Manousakis, G.; Lee, M.S. Progressive External Ophthalmoplegia. Curr. Neurol. Neurosci. Rep. 2016, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Majander, A.; Haltia, M.; Somer, H.; Lonnqvist, J.; Savontaus, M.L.; Peltonen, L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J. Clin. Investig. 1992, 90, 61–66. [Google Scholar] [CrossRef]
- Suomalainen, A.; Majander, A.; Wallin, M.; Setala, K.; Kontula, K.; Leinonen, H.; Salmi, T.; Paetau, A.; Haltia, M.; Valanne, L.; et al. Autosomal dominant progressive external ophthalmoplegia with multiple deletions of mtDNA: Clinical, biochemical, and molecular genetic features of the 10q-linked disease. Neurology 1997, 48, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.T.; Rosenberg, A.H.; Griffin, K.; Studier, F.W.; Patel, S.S. Biochemical analysis of mutant T7 primase/helicase proteins defective in DNA binding, nucleotide hydrolysis, and the coupling of hydrolysis with DNA unwinding. J. Biol. Chem. 1996, 271, 26825–26834. [Google Scholar] [CrossRef]
- Ariza, A.; Richard, D.J.; White, M.F.; Bond, C.S. Conformational flexibility revealed by the crystal structure of a crenarchaeal RadA. Nucleic Acids Res. 2005, 33, 1465–1473. [Google Scholar] [CrossRef]
- Zhang, Y.; Palla, M.; Sun, A.; Liao, J.C. Identification of unique interactions between the flexible linker and the RecA-like domains of DEAD-box helicase Mss116. J. Phys. Condens. Matter 2013, 25, 374101. [Google Scholar] [CrossRef]
- Matsushima, Y.; Farr, C.L.; Fan, L.; Kaguni, L.S. Physiological and biochemical defects in carboxyl-terminal mutants of mitochondrial DNA helicase. J Biol Chem 2008, 283, 23964–23971. [Google Scholar] [CrossRef]
- Matsushima, Y.; Kaguni, L.S. Functional importance of the conserved N-terminal domain of the mitochondrial replicative DNA helicase. Biochim. Biophys. Acta 2009, 1787, 290–295. [Google Scholar] [CrossRef][Green Version]
- GnomAD: genome aggregation database. Available online: https://gnomad.broadinstitute.org/ (accessed on 9 April 2020).
- Wanrooij, S.; Goffart, S.; Pohjoismaki, J.L.; Yasukawa, T.; Spelbrink, J.N. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Re.s 2007, 35, 3238–3251. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [PubMed]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lonnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Gulsuner, S.; Stapleton, G.A.; Walsh, T.; Lee, M.K.; Mandell, J.B.; Morales, A.; Klevit, R.E.; King, M.C.; Rogers, R.C. Infantile onset spinocerebellar ataxia caused by compound heterozygosity for Twinkle mutations and modeling of Twinkle mutations causing recessive disease. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001107. [Google Scholar] [CrossRef]
- Remtulla, S.; Emilie Nguyen, C.T.; Prasad, C.; Campbell, C. Twinkle-Associated Mitochondrial DNA Depletion. Pediatr. Neurol. 2019, 90, 61–65. [Google Scholar] [CrossRef]
- Sarzi, E.; Goffart, S.; Serre, V.; Chretien, D.; Slama, A.; Munnich, A.; Spelbrink, J.N.; Rotig, A. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann. Neurol. 2007, 62, 579–587. [Google Scholar] [CrossRef]
- Gottschalk, M.E.; Coker, S.B.; Fox, L.A. Neurologic anomalies of Perrault syndrome. Am. J. Med. Genet. 1996, 65, 274–276. [Google Scholar] [CrossRef]
- Perrault, M.; Klotz, B.; Housset, E. Two cases of Turner syndrome with deaf-mutism in two sisters. Bull. Mem. Soc. Med. Hop. Paris 1951, 67, 79–84. [Google Scholar]
- Fekete, B.; Pentelenyi, K.; Rudas, G.; Gal, A.; Grosz, Z.; Illes, A.; Idris, J.; Csukly, G.; Domonkos, A.; Molnar, M.J. Broadening the phenotype of the TWNK gene associated Perrault syndrome. BMC Med. Genet. 2019, 20, 198. [Google Scholar] [CrossRef]
- Morino, H.; Pierce, S.B.; Matsuda, Y.; Walsh, T.; Ohsawa, R.; Newby, M.; Hiraki-Kamon, K.; Kuramochi, M.; Lee, M.K.; Klevit, R.E.; et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 2014, 83, 2054–2061. [Google Scholar] [CrossRef]
- Futami, K.; Shimamoto, A.; Furuichi, Y. Mitochondrial and nuclear localization of human Pif1 helicase. Biol. Pharm. Bull. 2007, 30, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Kitao, S.; Ohsugi, I.; Ichikawa, K.; Goto, M.; Furuichi, Y.; Shimamoto, A. Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics 1998, 54, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Dmochowska, A.; Kalita, K.; Krawczyk, M.; Golik, P.; Mroczek, K.; Lazowska, J.; Stepien, P.P.; Bartnik, E. A human putative Suv3-like RNA helicase is conserved between Rhodobacter and all eukaryotes. Acta Biochim. Pol. 1999, 46, 155–162. [Google Scholar] [CrossRef] [PubMed]
- de Souza-Pinto, N.C.; Aamann, M.D.; Kulikowicz, T.; Stevnsner, T.V.; Bohr, V.A. Mitochondrial helicases and mitochondrial genome maintenance. Mech. Ageing Dev. 2010, 131, 503–510. [Google Scholar] [CrossRef]
- Szczesny, R.J.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.J.; Skrok, M.M.; Golik, P.; Stepien, P.P. Yeast and human mitochondrial helicases. Biochim Biophys Acta 2013, 1829, 842–853. [Google Scholar] [CrossRef]
- Boule, J.B.; Zakian, V.A. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Res. 2006, 34, 4147–4153. [Google Scholar] [CrossRef]
- Zhang, D.H.; Zhou, B.; Huang, Y.; Xu, L.X.; Zhou, J.Q. The human Pif1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res. 2006, 34, 1393–1404. [Google Scholar] [CrossRef]
- Byrd, A.K.; Raney, K.D. Structure and function of Pif1 helicase. Biochem. Soc. Trans. 2017, 45, 1159–1171. [Google Scholar] [CrossRef]
- Bochman, M.L.; Sabouri, N.; Zakian, V.A. Unwinding the functions of the Pif1 family helicases. DNA Repair 2010, 9, 237–249. [Google Scholar] [CrossRef]
- Kazak, L.; Reyes, A.; Duncan, A.L.; Rorbach, J.; Wood, S.R.; Brea-Calvo, G.; Gammage, P.A.; Robinson, A.J.; Minczuk, M.; Holt, I.J. Alternative translation initiation augments the human mitochondrial proteome. Nucleic Acids Res. 2013, 41, 2354–2369. [Google Scholar] [CrossRef]
- Snow, B.E.; Mateyak, M.; Paderova, J.; Wakeham, A.; Iorio, C.; Zakian, V.; Squire, J.; Harrington, L. Murine Pif1 interacts with telomerase and is dispensable for telomere function in vivo. Mol. Cell Biol. 2007, 27, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- George, T.; Wen, Q.; Griffiths, R.; Ganesh, A.; Meuth, M.; Sanders, C.M. Human Pif1 helicase unwinds synthetic DNA structures resembling stalled DNA replication forks. Nucleic Acids Res. 2009, 37, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.M. Human Pif1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem. J. 2010, 430, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.A.; Singh, S.P.; Burgers, P.M.; Galletto, R. Complementary roles of Pif1 helicase and single stranded DNA binding proteins in stimulating DNA replication through G-quadruplexes. Nucleic Acids Res. 2019, 47, 8595–8605. [Google Scholar] [CrossRef]
- Butler, T.J.; Estep, K.N.; Sommers, J.A.; Maul, R.W.; Moore, A.Z.; Bandinelli, S.; Cucca, F.; Tuke, M.A.; Wood, A.R.; Bharti, S.K.; et al. Mitochondrial genetic variation is enriched in G-quadruplex regions that stall DNA synthesis in vitro. Hum. Mol. Genet. 2020. [Google Scholar] [CrossRef]
- Bannwarth, S.; Berg-Alonso, L.; Auge, G.; Fragaki, K.; Kolesar, J.E.; Lespinasse, F.; Lacas-Gervais, S.; Burel-Vandenbos, F.; Villa, E.; Belmonte, F.; et al. Inactivation of Pif1 helicase causes a mitochondrial myopathy in mice. Mitochondrion 2016, 30, 126–137. [Google Scholar] [CrossRef]
- Bae, S.H.; Seo, Y.S. Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J. Biol. Chem. 2000, 275, 38022–38031. [Google Scholar] [CrossRef]
- Budd, M.E.; Choe, W.; Campbell, J.L. The nuclease activity of the yeast DNA2 protein, which is related to the RecB-like nucleases, is essential in vivo. J. Biol. Chem. 2000, 275, 16518–16529. [Google Scholar] [CrossRef]
- Budd, M.E.; Choe, W.C.; Campbell, J.L. DNA2 encodes a DNA helicase essential for replication of eukaryotic chromosomes. J. Biol. Chem. 1995, 270, 26766–26769. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, D.W.; Bae, S.H.; Kim, J.A.; Ryu, G.H.; Kwon, Y.N.; Kim, K.A.; Koo, H.S.; Seo, Y.S. The endonuclease activity of the yeast Dna2 enzyme is essential in vivo. Nucleic Acids Res. 2000, 28, 2873–2881. [Google Scholar] [CrossRef]
- Imamura, O.; Campbell, J.L. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast dna2 mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 8193–8198. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef]
- Ronchi, D.; Di Fonzo, A.; Lin, W.; Bordoni, A.; Liu, C.; Fassone, E.; Pagliarani, S.; Rizzuti, M.; Zheng, L.; Filosto, M.; et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 2013, 92, 293–300. [Google Scholar] [CrossRef]
- Jia, P.P.; Junaid, M.; Ma, Y.B.; Ahmad, F.; Jia, Y.F.; Li, W.G.; Pei, D.S. Role of human DNA2 (hDNA2) as a potential target for cancer and other diseases: A systematic review. DNA Repair 2017, 59, 9–19. [Google Scholar] [CrossRef]
- Suzuki, T.; Kohno, T.; Ishimi, Y. DNA helicase activity in purified human RECQL4 protein. J. Biochem. 2009, 146, 327–335. [Google Scholar] [CrossRef]
- Croteau, D.L.; Rossi, M.L.; Canugovi, C.; Tian, J.; Sykora, P.; Ramamoorthy, M.; Wang, Z.M.; Singh, D.K.; Akbari, M.; Kasiviswanathan, R.; et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 2012, 11, 456–466. [Google Scholar] [CrossRef]
- De, S.; Kumari, J.; Mudgal, R.; Modi, P.; Gupta, S.; Futami, K.; Goto, H.; Lindor, N.M.; Furuichi, Y.; Mohanty, D.; et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J. Cell Sci. 2012, 125, 2509–2522. [Google Scholar] [CrossRef]
- Keller, H.; Kiosze, K.; Sachsenweger, J.; Haumann, S.; Ohlenschlager, O.; Nuutinen, T.; Syvaoja, J.E.; Gorlach, M.; Grosse, F.; Pospiech, H. The intrinsically disordered amino-terminal region of human RecQL4: Multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic Acids Res. 2014, 42, 12614–12627. [Google Scholar] [CrossRef]
- Marino, F.; Mojumdar, A.; Zucchelli, C.; Bhardwaj, A.; Buratti, E.; Vindigni, A.; Musco, G.; Onesti, S. Structural and biochemical characterization of an RNA/DNA binding motif in the N-terminal domain of RecQ4 helicases. Sci. Rep. 2016, 6, 21501. [Google Scholar] [CrossRef] [PubMed]
- Sedlackova, H.; Cechova, B.; Mlcouskova, J.; Krejci, L. RECQ4 selectively recognizes Holliday junctions. DNA Repair 2015, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009, 28, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Nie, L.; Peng, Z.; Yang, Q.; Yang, K.; Tao, J.; Mi, Y.; Fang, X.; Balajee, A.S.; Zhao, Y. RecQL4 cytoplasmic localization: Implications in mitochondrial DNA oxidative damage repair. Int. J. Biochem. Cell Biol. 2012, 44, 1942–1951. [Google Scholar] [CrossRef]
- Wang, J.T.; Xu, X.; Alontaga, A.Y.; Chen, Y.; Liu, Y. Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 2014, 7, 848–858. [Google Scholar] [CrossRef]
- Bohr, V.A. Rising from the RecQ-age: The role of human RecQ helicases in genome maintenance. Trends Biochem. Sci. 2008, 33, 609–620. [Google Scholar] [CrossRef]
- Wu, J.; Capp, C.; Feng, L.; Hsieh, T.S. Drosophila homologue of the Rothmund-Thomson syndrome gene: Essential function in DNA replication during development. Dev. Biol. 2008, 323, 130–142. [Google Scholar] [CrossRef]
- Minczuk, M.; Dmochowska, A.; Palczewska, M.; Stepien, P.P. Overexpressed yeast mitochondrial putative RNA helicase Mss116 partially restores proper mtRNA metabolism in strains lacking the Suv3 mtRNA helicase. Yeast 2002, 19, 1285–1293. [Google Scholar] [CrossRef]
- Szczesny, R.J.; Obriot, H.; Paczkowska, A.; Jedrzejczak, R.; Dmochowska, A.; Bartnik, E.; Formstecher, P.; Polakowska, R.; Stepien, P.P. Down-regulation of human RNA/DNA helicase SUV3 induces apoptosis by a caspase- and AIF-dependent pathway. Biol. Cell 2007, 99, 323–332. [Google Scholar] [CrossRef]
- Wang, Y.; Bogenhagen, D.F. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 2006, 281, 25791–25802. [Google Scholar] [CrossRef]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2010, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Guo, X.E.; Huang, C.K.; Shew, J.Y.; Reddick, R.L.; Wallace, D.C.; Lee, W.H. Mitochondrial genome instability resulting from SUV3 haploinsufficiency leads to tumorigenesis and shortened lifespan. Oncogene 2013, 32, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Khidr, L.; Wu, G.; Davila, A.; Procaccio, V.; Wallace, D.; Lee, W.H. Role of SUV3 helicase in maintaining mitochondrial homeostasis in human cells. J. Biol. Chem. 2008, 283, 27064–27073. [Google Scholar] [CrossRef] [PubMed]
- Valgardsdottir, R.; Brede, G.; Eide, L.G.; Frengen, E.; Prydz, H. Cloning and characterization of MDDX28, a putative dead-box helicase with mitochondrial and nuclear localization. J. Biol. Chem. 2001, 276, 32056–32063. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.J.; Tsukahara, M.; Liu, E.; Ye, L.; Xiong, H.; Noguchi, S.; Suzuki, K.; Ji, Z.S. The novel helicase helG (DHX30) is expressed during gastrulation in mice and has a structure similar to a human DExH box helicase. Stem. Cells Dev. 2015, 24, 372–383. [Google Scholar] [CrossRef]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef]
- Tu, Y.T.; Barrientos, A. The Human Mitochondrial DEAD-Box Protein DDX28 Resides in RNA Granules and Functions in Mitoribosome Assembly. Cell Rep. 2015, 10, 854–864. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, B.; Falkenberg, M. TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes 2020, 11, 408. https://doi.org/10.3390/genes11040408
Peter B, Falkenberg M. TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes. 2020; 11(4):408. https://doi.org/10.3390/genes11040408
Chicago/Turabian StylePeter, Bradley, and Maria Falkenberg. 2020. "TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease" Genes 11, no. 4: 408. https://doi.org/10.3390/genes11040408
APA StylePeter, B., & Falkenberg, M. (2020). TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes, 11(4), 408. https://doi.org/10.3390/genes11040408