Genomics of MPNST (GeM) Consortium: Rationale and Study Design for Multi-Omic Characterization of NF1-Associated and Sporadic MPNSTs

 , ,
, ,  , , ,
, , ,
Abstract
1. The Complex Genomic Landscape of MPNST (Malignant Peripheral Nerve Sheath Tumors)
2. Knowledge Gaps in the Understanding of Tumor Drivers and Evolution of MPNST
3. Establishment of the Genomics of MPNST Consortium to Address Knowledge Gaps
4. Specimen and Clinical Data Collection, and Specimen Processing
5. Plan for Multi-Omic Characterization of MPNST
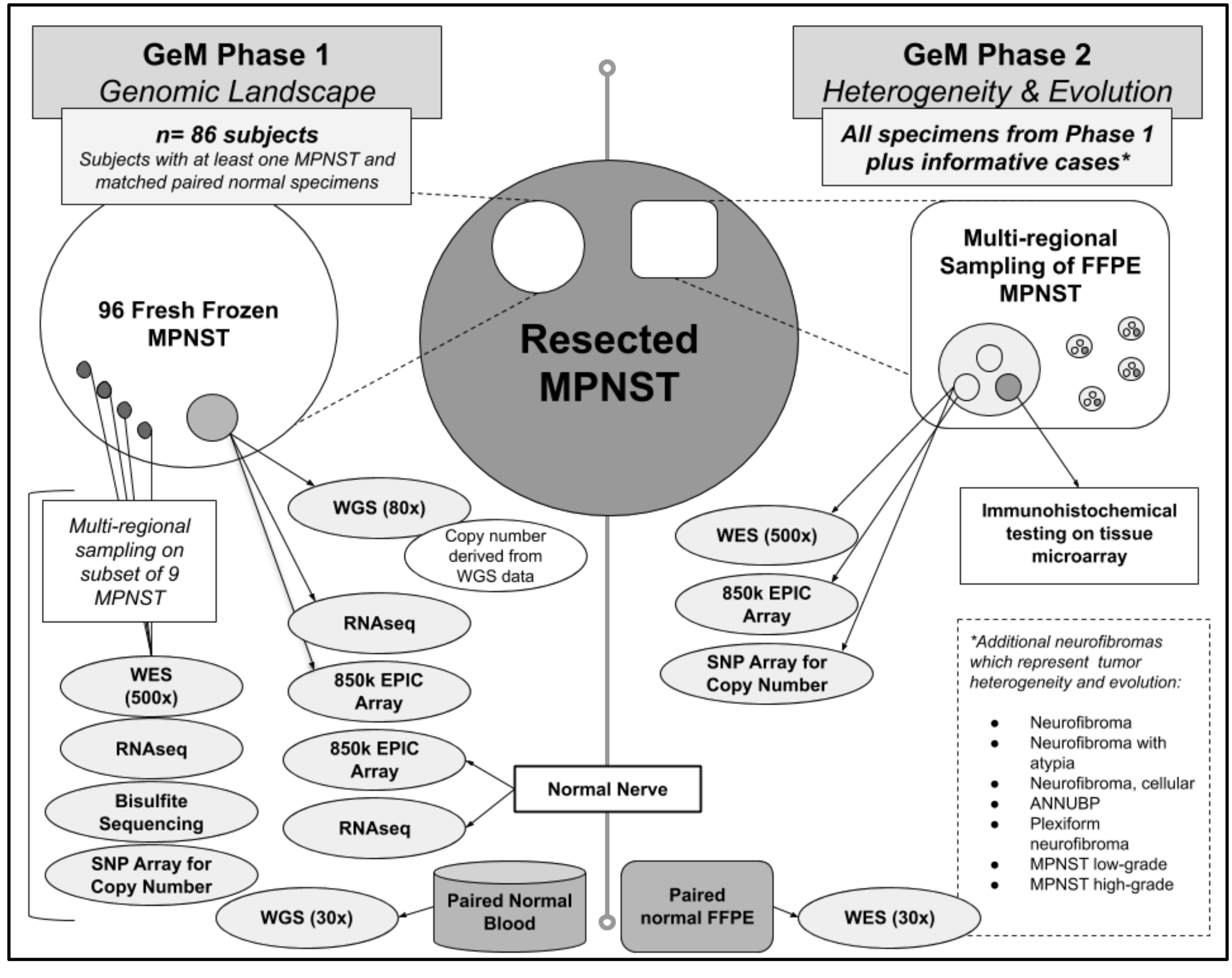
5.1. Phase 1: Multi-Omic Profiling on Frozen Tumor Material to Study MPNST Genomic Complexity, Tumor Drivers, and Tumor Heterogeneity
5.2. Phase 2: Extensive Characterization of Tumor Heterogeneity and Evolution Using FFPE MPNST Samples from Phase 1 and Additional Informative Cases
5.3. Bioinformatics Analysis for Multi-Omic Profiling of MPNSTs
5.4. GeM Data Display and Availability
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Evans, D.G.; Huson, S.M.; Birch, J.M. Malignant peripheral nerve sheath tumours in inherited disease. Clin. Sarcoma Res. 2012, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Spiliopoulos, K.; Plotkin, S.R.; Hornicek, F.J.; Harmon, D.C.; Delaney, T.F.; Williams, Z. Role of resection of malignant peripheral nerve sheath tumors in patients with neurofibromatosis type 1. J. Neurosurg. 2013, 118, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Shiu, M.H.; Senie, R.T.; Woodruff, J.M. Malignant peripheral nerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer 1990, 66, 1253–1265. [Google Scholar] [CrossRef]
- Frustaci, S.; Gherlinzoni, F.; De Paoli, A.; Bonetti, M.; Azzarelli, A.; Comandone, A.; Olmi, P.; Buonadonna, A.; Pignatti, G.; Barbieri, E.; et al. Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: Results of the Italian randomized cooperative trial. J. Clin. Oncol. 2001, 19, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, A.; Citrin, D. The role of radiation therapy in the management of sarcomas. Surg. Clin. N. Am. 2008, 88, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Chang, A.E.; Baker, A.R.; Sindelar, W.F.; Danforth, D.N.; Topalian, S.L.; DeLaney, T.; Glatstein, E.; Steinberg, S.M.; Merino, M.J.; et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J. Clin. Oncol. 1998, 16, 197–203. [Google Scholar] [CrossRef]
- Ferner, R.E.; Gutmann, D.H. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002, 62, 1573–1577. [Google Scholar]
- Prudner, B.C.; Ball, T.; Rathore, R.; Hirbe, A.C. Diagnosis and management of malignant peripheral nerve sheath tumors: Current practice and future perspectives. Neuro Oncol. Adv. 2019. [Google Scholar] [CrossRef]
- Kim, A.; Stewart, D.R.; Reilly, K.M.; Viskochil, D.; Miettinen, M.M.; Widemann, B.C. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma 2017, 2017, 7429697. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef] [PubMed]
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of human malignant peripheral nerve sheath tumors. Neuro Oncol. Adv. 2019. [Google Scholar] [CrossRef]
- Rohrich, M.; Koelsche, C.; Schrimpf, D.; Capper, D.; Sahm, F.; Kratz, A.; Reuss, J.; Hovestadt, V.; Jones, D.T.; Bewerunge-Hudler, M.; et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016, 131, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, S.A.; Lind, G.E.; Kolberg, M.; Holand, M.; Bjerkehagen, B.; Sundby Hall, K.; van den Berg, E.; Mertens, F.; Smeland, S.; Picci, P.; et al. Methylated RASSF1A in malignant peripheral nerve sheath tumors identifies neurofibromatosis type 1 patients with inferior prognosis. Neuro Oncol. 2015, 17, 63–69. [Google Scholar] [CrossRef]
- Pemov, A.; Li, H.; Patidar, R.; Hansen, N.F.; Sindiri, S.; Hartley, S.W.; Wei, J.S.; Elkahloun, A.; Chandrasekharappa, S.C.; Program, N.C.S.; et al. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene 2017, 36, 3168–3177. [Google Scholar] [CrossRef]
- Eisenbarth, I.; Beyer, K.; Krone, W.; Assum, G. Toward a survey of somatic mutation of the NF1 gene in benign neurofibromas of patients with neurofibromatosis type 1. Am. J. Hum. Genet. 2000, 66, 393–401. [Google Scholar] [CrossRef]
- Carroll, S.L. The Challenge of Cancer Genomics in Rare Nervous System Neoplasms: Malignant Peripheral Nerve Sheath Tumors as a Paradigm for Cross-Species Comparative Oncogenomics. Am. J. Pathol. 2016, 186, 464–477. [Google Scholar] [CrossRef]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef]
- Wu, L.M.N.; Deng, Y.; Wang, J.; Zhao, C.; Wang, J.; Rao, R.; Xu, L.; Zhou, W.; Choi, K.; Rizvi, T.A.; et al. Programming of Schwann Cells by Lats1/2-TAZ/YAP Signaling Drives Malignant Peripheral Nerve Sheath Tumorigenesis. Cancer Cell 2018, 33, 292–308. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Zambo, I.; Vesely, K. WHO classification of tumours of soft tissue and bone 2013: The main changes compared to the 3rd edition. Cesk. Patol. 2014, 50, 64–70. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Electronic address, E.D.S.C.; Cancer Genome Atlas Research, N. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, S.; Park, W.Y.; Kim, T.M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Steele, C.D.; Tarabichi, M.; Oukrif, D.; Webster, A.P.; Ye, H.; Fittall, M.; Lombard, P.; Martincorena, I.; Tarpey, P.S.; Collord, G.; et al. Undifferentiated Sarcomas Develop through Distinct Evolutionary Pathways. Cancer Cell 2019, 35, 441–456. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| NF1-Related | Sporadic or Unknown Diagnosis | Total MPNST | |
|---|---|---|---|
| n (%) | n (%) | n (%) | |
| Fresh Frozen MPNST | 60 (62.5%) | 36 (39.6%) | 96 (100%) |
| Tumor Grade | |||
| Low Grade | 5 (5.2%) | 2 (2.1%) | 7 (7.3%) |
| High Grade | 49 (51.0%) | 32 (33.3%) | 81 (84.4%) |
| Unknown | 6 (6.3%) | 2 (2.1%) | 8 (8.3%) |
| Neo-Adjuvant Treatment | |||
| Chemotherapy | 8 (8.3%) | 5 (5.2%) | 13 (13.5%) |
| Radiation | 3 (3.1%) | 9 (9.4%) | 12 (12.5%) |
| Chemotherapy and Radiation | 2 (2.1%) | 2 (2.1%) | 4 (4.2%) |
| No neo-adjuvant treatment | 44 (45.8%) | 20 (20.8%) | 64 (66.7%) |
| Unknown | 3 (3.1%) | 0 (0%) | 3 (3.1%) |
| Tumor Anatomic Location | |||
| Head/Neck/Face | 4 (4.2%) | 0 (0%) | 4 (4.2%) |
| Lower Limb | 18 (18.8%) | 18 (18.8%) | 36 (37.5%) |
| Upper Limb | 14 (14.6%) | 12 (12.5%) | 26 (27.1%) |
| Brachial Plexus | 3 (3.1%) | 1 (1.0%) | 4 (4.2%) |
| Lumbosacral Plexus | 5 (5.2%) | 3 (3.1%) | 8 (8.3%) |
| Trunk | 8 (8.3%) | 1 (1.0%) | 9 (9.4%) |
| Retroperitoneum | 1 (1.0%) | 0 (0%) | 1 (1.0%) |
| Other | 7 (7.3%) | 1 (1.0%) | 8 (8.3%) |
| Total MPNST | 60 (62.5%) | 36 (37.5%) | 96 (100%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, D.T.; Cortés-Ciriano, I.; Pillay, N.; Hirbe, A.C.; Snuderl, M.; Bui, M.M.; Piculell, K.; Al-Ibraheemi, A.; Dickson, B.C.; Hart, J.; et al. Genomics of MPNST (GeM) Consortium: Rationale and Study Design for Multi-Omic Characterization of NF1-Associated and Sporadic MPNSTs. Genes 2020, 11, 387. https://doi.org/10.3390/genes11040387
Miller DT, Cortés-Ciriano I, Pillay N, Hirbe AC, Snuderl M, Bui MM, Piculell K, Al-Ibraheemi A, Dickson BC, Hart J, et al. Genomics of MPNST (GeM) Consortium: Rationale and Study Design for Multi-Omic Characterization of NF1-Associated and Sporadic MPNSTs. Genes. 2020; 11(4):387. https://doi.org/10.3390/genes11040387
Chicago/Turabian StyleMiller, David T., Isidro Cortés-Ciriano, Nischalan Pillay, Angela C. Hirbe, Matija Snuderl, Marilyn M. Bui, Katherine Piculell, Alyaa Al-Ibraheemi, Brendan C. Dickson, Jesse Hart, and et al. 2020. "Genomics of MPNST (GeM) Consortium: Rationale and Study Design for Multi-Omic Characterization of NF1-Associated and Sporadic MPNSTs" Genes 11, no. 4: 387. https://doi.org/10.3390/genes11040387
APA StyleMiller, D. T., Cortés-Ciriano, I., Pillay, N., Hirbe, A. C., Snuderl, M., Bui, M. M., Piculell, K., Al-Ibraheemi, A., Dickson, B. C., Hart, J., Jones, K., Jordan, J. T., Kim, R. H., Lindsay, D., Nishida, Y., Ullrich, N. J., Wang, X., Park, P. J., & Flanagan, A. M., on behalf of the Genomics of MPNST (GeM) Consortium. (2020). Genomics of MPNST (GeM) Consortium: Rationale and Study Design for Multi-Omic Characterization of NF1-Associated and Sporadic MPNSTs. Genes, 11(4), 387. https://doi.org/10.3390/genes11040387