Homozygous Splice Site Mutation in ZP1 Causes Familial Oocyte Maturation Defect

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
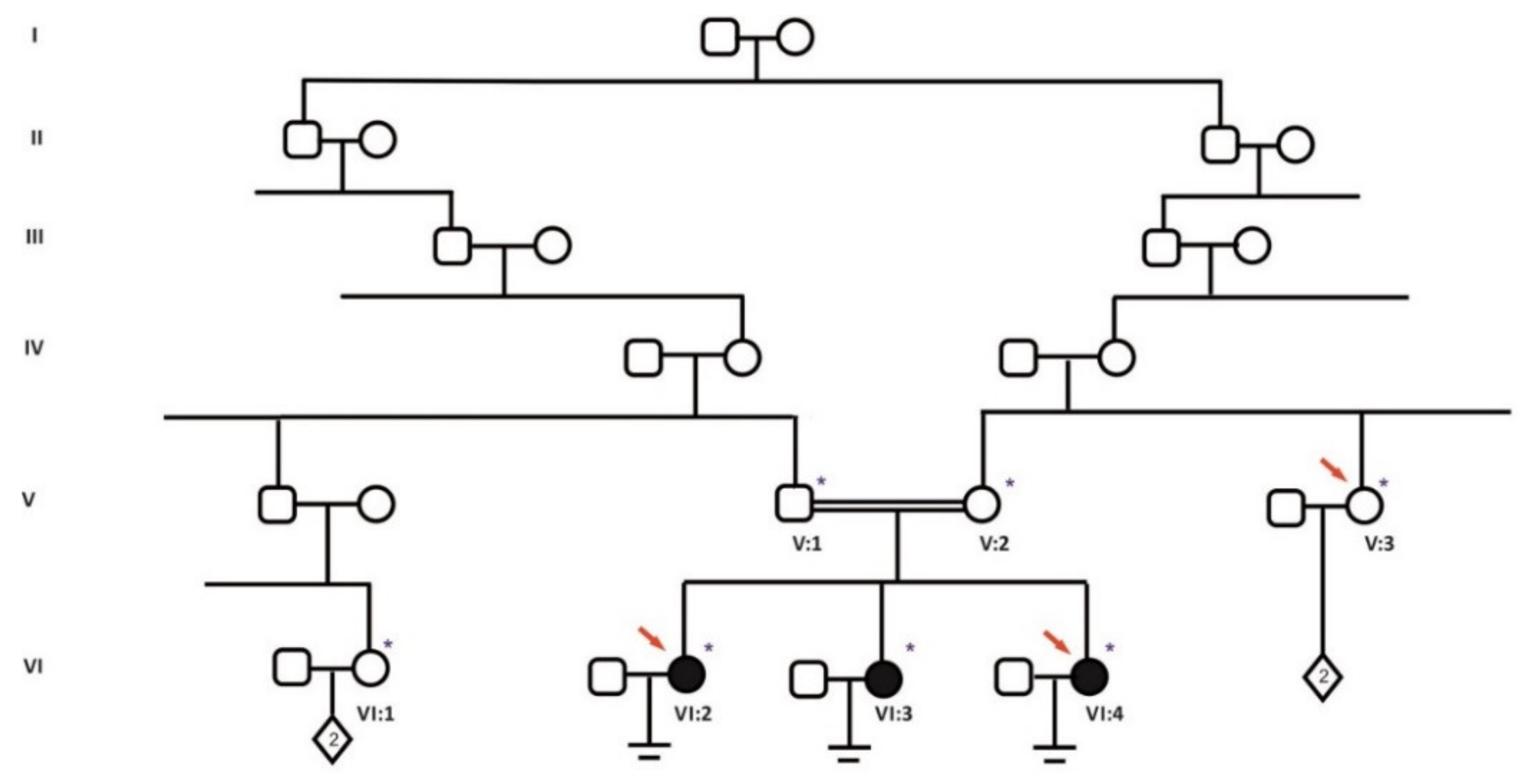
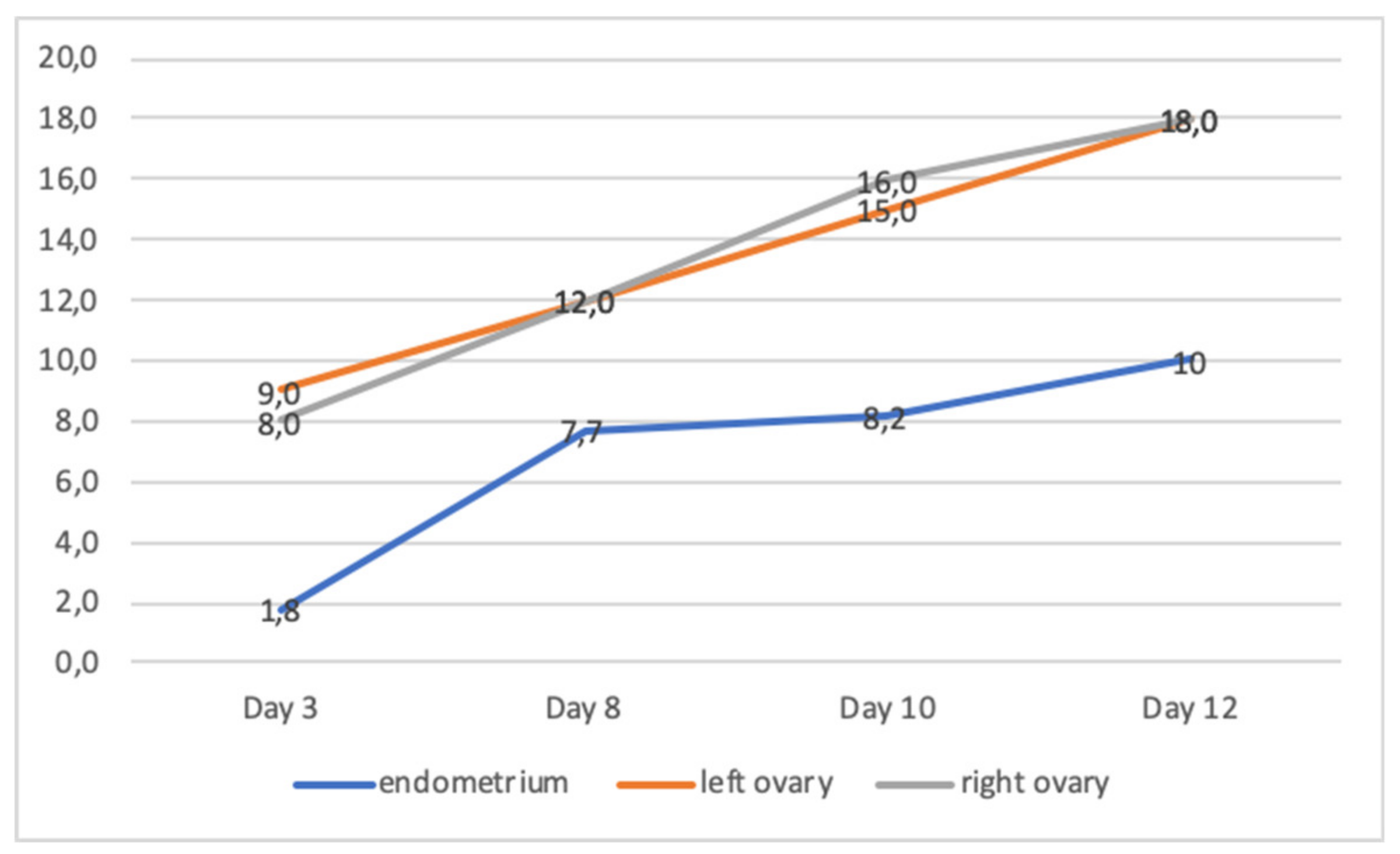
2.1. Patients and Collected Samples
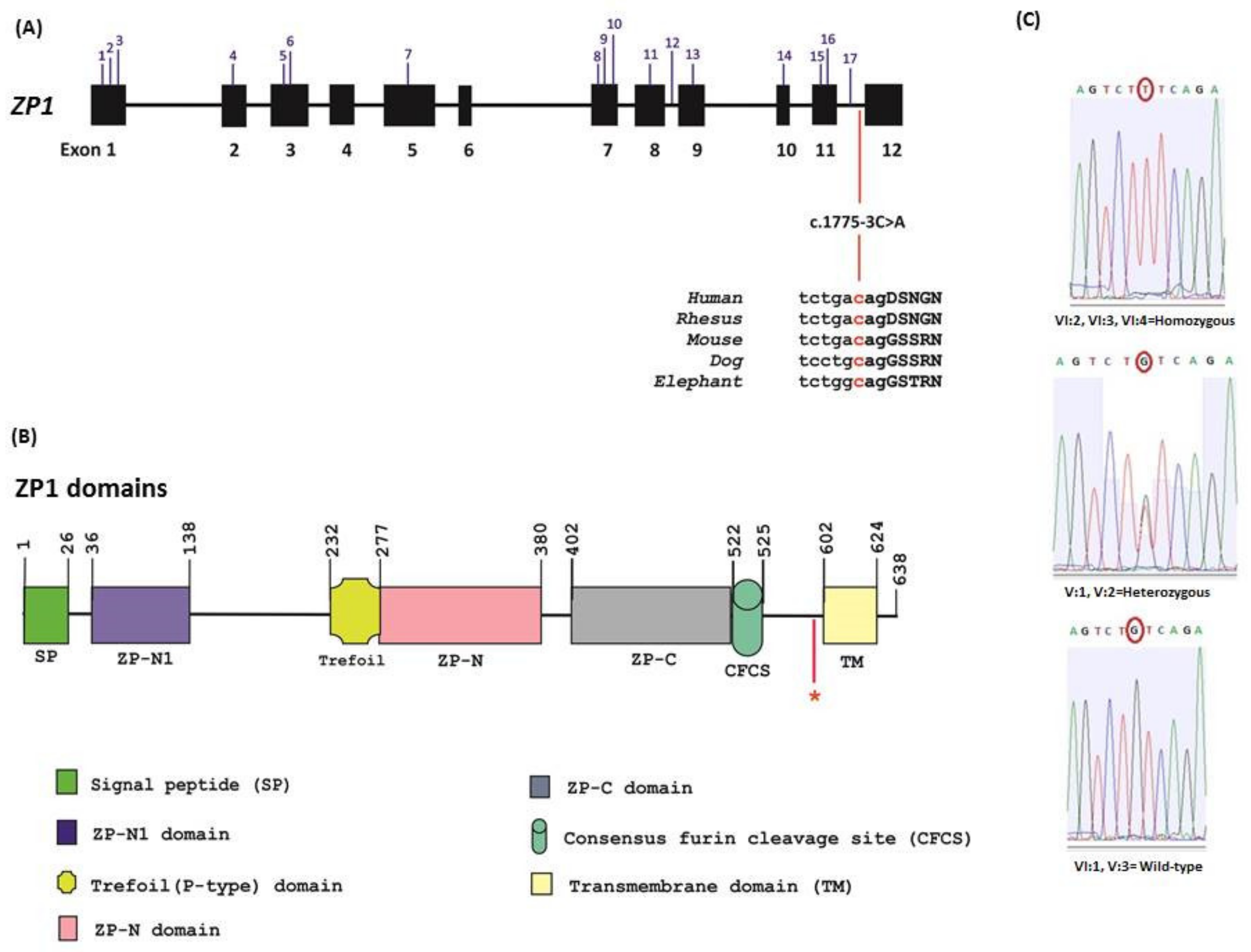
2.2. Whole Exome Sequencing and Data Analysis
2.3. Mutation Screening
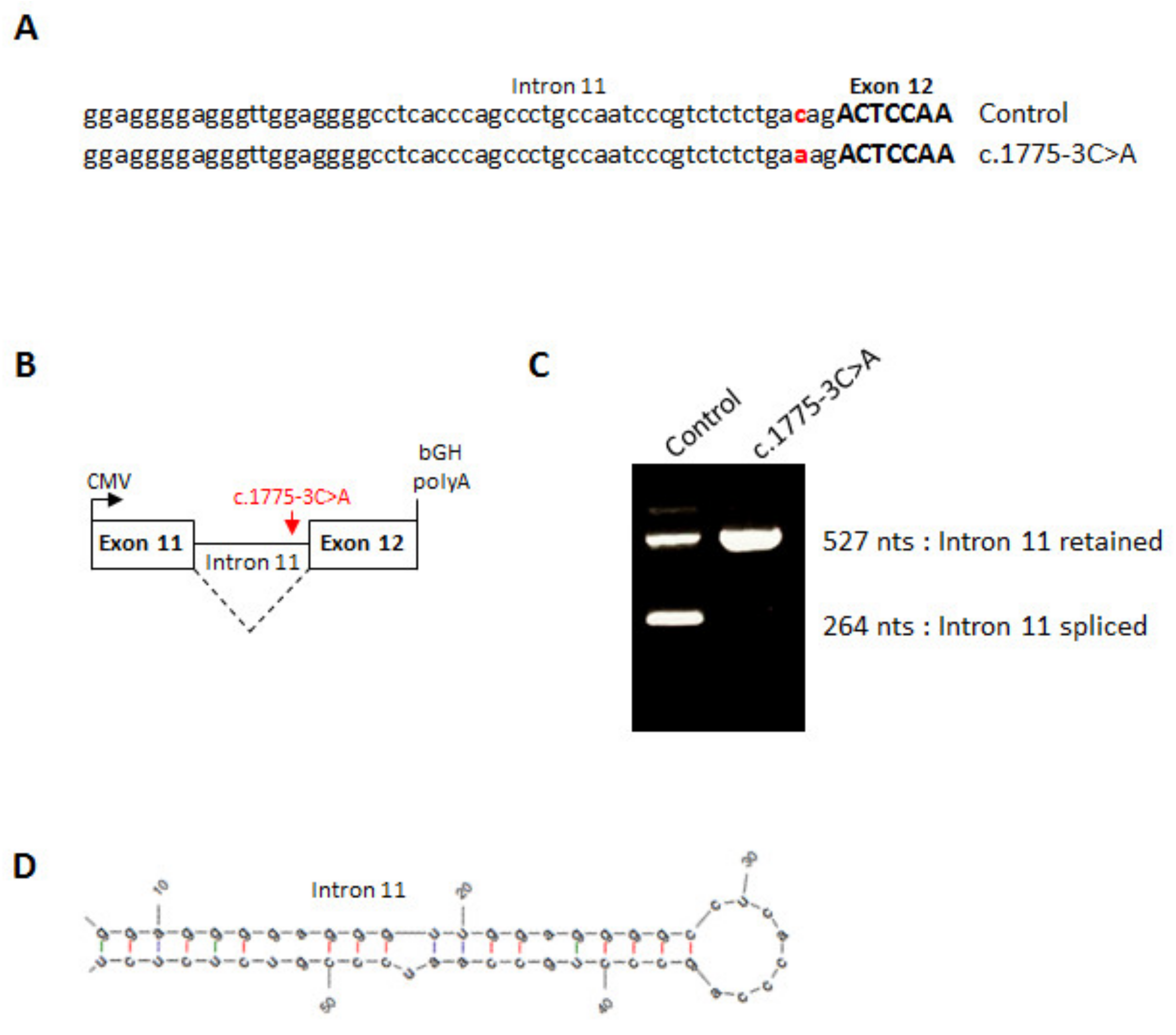
2.4. Minigene Assay
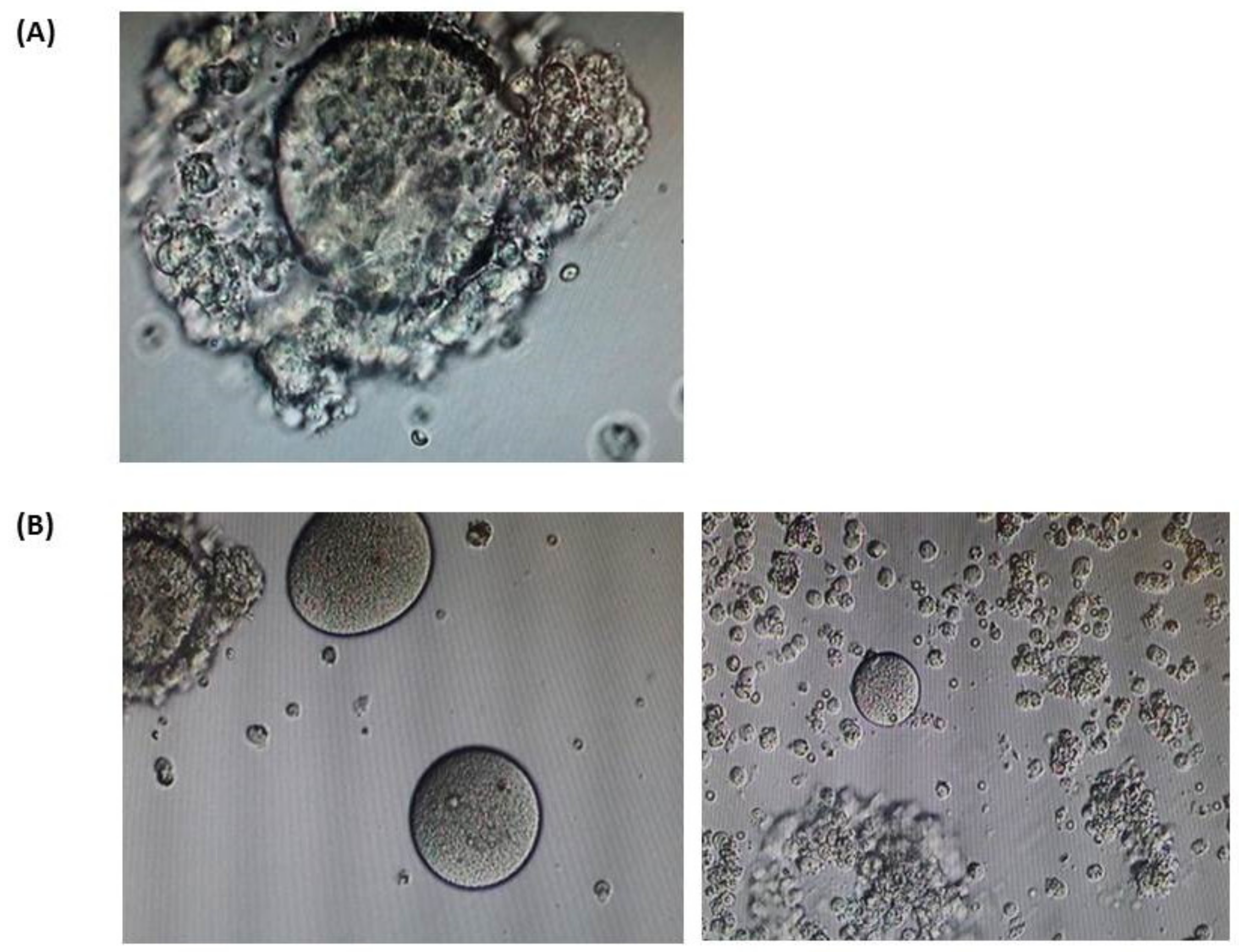
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coticchio, G.; Dal Canto, M.; Mignini Renzini, M.; Guglielmo, M.C.; Brambillasca, F.; Turchi, D.; Novara, P.V.; Fadini, R. Oocyte maturation: Gamete-somatic cells interactions, meiotic resumption, cytoskeletal dynamics and cytoplasmic reorganization. Hum. Reprod. Update 2015, 21, 427–454. [Google Scholar] [CrossRef] [PubMed]
- Mehlmann, L.M. Stops and starts in mammalian oocytes: Recent advances in understanding the regulation of meiotic arrest and oocyte maturation. Reproduction 2005, 130, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Eppig, J.J.; O’Brien, M.; Wigglesworth, K. Mammalian oocyte growth and development in vitro. Mol. Reprod. Dev. 1996, 44, 260–273. [Google Scholar] [CrossRef]
- Dean, J. Exacting requirements for development of the egg. N. Engl. J. Med. 2016, 374, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Sang, Q.; Kuang, Y.; Sun, X.; Yan, Z.; Zhang, S.; Shi, J.; Tian, G.; Luchniak, A.; Fukuda, Y.; et al. Mutations in TUBB8 and human oocyte meiotic arrest. N. Engl. J. Med. 2016, 374, 223–232. [Google Scholar] [CrossRef]
- Feng, R.; Yan, Z.; Li, B.; Yu, M.; Sang, Q.; Tian, G.; Xu, Y.; Chen, B.; Qu, R.; Sun, Z.; et al. Mutations in TUBB8 cause a multiplicity of phenotypes in human oocytes and early embryos. J. Med. Genet. 2016, 53, 662–671. [Google Scholar] [CrossRef]
- Huang, L.; Tong, X.; Luo, L.; Zheng, S.; Jin, R.; Fu, Y.; Zhou, G.; Li, D.; Liu, Y. Mutation analysis of the TUBB8 gene in nine infertile women with oocyte maturation arrest. Reprod. Biomed. Online 2017, 35, 305–310. [Google Scholar] [CrossRef]
- Chen, B.; Li, B.; Li, D.; Yan, Z.; Mao, X.; Xu, Y.; Mu, J.; Li, Q.; Jin, L.; He, L.; et al. Novel mutations and structural deletions in TUBB8: Expanding mutational and phenotypic spectrum of patients with arrest in oocyte maturation, fertilization or early embryonic development. Hum. Reprod. 2017, 32, 457–464. [Google Scholar] [CrossRef]
- Wang, A.C.; Zhang, Y.S.; Wang, B.S.; Zhao, X.Y.; Wu, F.X.; Zhai, X.H.; Sun, J.X.; Mei, S.Y. Mutation analysis of the TUBB8 gene in primary infertile women with arrest in oocyte maturation. Gynecol. Endocrinol. 2018, 34, 900–904. [Google Scholar] [CrossRef]
- Yuan, P.; Zheng, L.; Liang, H.; Li, Y.; Zhao, H.; Li, R.; Lai, L.; Zhang, Q.; Wang, W. A novel mutation in the TUBB8 gene is associated with complete cleavage failure in fertilized eggs. J. Assist. Reprod. Genet. 2018, 35, 1349–1356. [Google Scholar] [CrossRef]
- Xiang, J.; Wang, W.; Qian, C.; Xue, J.; Wang, T.; Li, H.; Li, H. Human oocyte maturation arrest caused by a novel missense mutation in TUBB8. J. Int. Med. Res. 2018, 46, 3759–3764. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, Z.; Sun, X.; Kuang, Y.; Mao, X.; Wang, X.; Yan, Z.; Li, B.; Xu, Y.; Yu, M.; et al. Biallelic mutations in PATL2 cause female infertility characterized by oocyte maturation arrest. Am. J. Hum. Genet. 2017, 101, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Coskun, S.; Alhassan, S.; Elnour, A.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Arold, S.T.; Alkuraya, F.S. Female infertility caused by mutations in the oocyte-specific translational repressor PATL2. Am. J. Hum. Genet. 2017, 101, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tong, X.; Wang, F.; Luo, L.; Jin, R.; Fu, Y.; Zhou, G.; Li, D.; Song, G.; Liu, Y.; et al. Novel mutations in PATL2 cause female infertility with oocyte germinal vesicle arrest. Hum. Reprod. 2018, 33, 1183–1190. [Google Scholar] [CrossRef]
- Wu, L.; Chen, H.; Li, D.; Song, D.; Chen, B.; Yan, Z.; Lyu, Q.; Wang, L.; Kuang, Y.; Li, B.; et al. Novel mutations in PATL2: Expanding the mutational spectrum and corresponding phenotypic variability associated with female infertility. J. Hum. Genet. 2019, 64, 379–385. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, L.; Wang, J.; Luo, G.; Xi, Q.; Zhou, X.; Li, Z.; Yang, X.; Duan, J.; Jin, L.; et al. Novel homozygous mutations in PATL2 lead to female infertility with oocyte maturation arrest. J. Assist. Reprod. Genet. 2020. [Google Scholar] [CrossRef]
- Sang, Q.; Li, B.; Kuang, Y.; Wang, X.; Zhang, Z.; Chen, B.; Wu, L.; Lyu, Q.; Fu, Y.; Yan, Z.; et al. Homozygous mutations in WEE2 cause fertilization failure and female infertility. Am. J. Hum. Genet. 2018, 102, 649–657. [Google Scholar] [CrossRef]
- Yang, X.; Shu, L.; Cai, L.; Sun, X.; Cui, Y.; Liu, J. Homozygous missense mutation Arg207Cys in the WEE2 gene causes female infertility and fertilization failure. J. Assist. Reprod. Genet. 2019, 36, 965–971. [Google Scholar] [CrossRef]
- Dai, J.; Zheng, W.; Dai, C.; Guo, J.; Lu, C.; Gong, F.; Li, Y.; Zhou, Q.; Lu, G.; Lin, G. New biallelic mutations in WEE2: Expanding the spectrum of mutations that cause fertilization failure or poor fertilization. Fertil. Steril. 2019, 111, 510–518. [Google Scholar] [CrossRef]
- Zhao, S.; Chen, T.; Yu, M.; Bian, Y.; Cao, Y.; Ning, Y.; Su, S.; Zhang, J.; Zhao, S. Novel WEE2 gene variants identified in patients with fertilization failure and female infertility. Fertil. Steril. 2019, 111, 519–526. [Google Scholar] [CrossRef]
- Zhou, X.; Zhu, L.; Hou, M.; Wu, Y.; Li, Z.; Wang, J.; Liu, Z.; Zhang, D.; Jin, L.; Zhang, X. Novel compound heterozygous mutations in WEE2 causes female infertility and fertilization failure. J. Assist. Reprod. Genet. 2019, 36, 1957–1962. [Google Scholar] [CrossRef]
- Sang, Q.; Zhang, Z.; Shi, J.; Sun, X.; Li, B.; Yan, Z.; Xue, S.; Ai, A.; Lyu, Q.; Li, W.; et al. A pannexin 1 channelopathy causes human oocyte death. Sci. Transl. Med. 2019, 11, eaav8731. [Google Scholar] [CrossRef]
- Yang, P.; Luan, X.; Peng, Y.; Chen, T.; Su, S.; Zhang, C.; Wang, Z.; Cheng, L.; Zhang, X.; Wang, Y.; et al. Novel zona pellucida gene variants identified in patients with oocyte anomalies. Fertil. Steril. 2017, 107, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Lv, C.; Zhao, Y.C.; Li, W.; He, X.M.; Li, P.; Sha, A.G.; Tian, X.; Papasian, C.J.; Deng, H.W.; et al. Mutant ZP1 in familial infertility. N. Engl. J. Med. 2014, 370, 1220–1226. [Google Scholar] [CrossRef]
- Zhou, Z.; Ni, C.; Wu, L.; Chen, B.; Xu, Y.; Zhang, Z.; Mu, J.; Li, B.; Yan, Z.; Fu, J.; et al. Novel mutations in ZP1, ZP2, and ZP3 cause female infertility due to abnormal zona pellucida formation. Hum. Genet. 2019, 138, 327–337. [Google Scholar] [CrossRef]
- Yuan, P.; Li, R.; Li, D.; Zheng, L.; Ou, S.; Zhao, H.; Zhang, Q.; Wang, W. Novel mutation in the ZP1 gene and clinical implications. J. Assist. Reprod. Genet. 2019, 36, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Chen, Y.; Hu, L.; Du, J.; Gong, F.; Dai, J.; Zhang, S.; Wang, M.; Chen, J.; Guo, J.; et al. ZP1 mutations are associated with empty follicle syndrome: Evidence for the existence of an intact oocyte and a zona pellucida in follicles up to the early antral stage. A case report. Hum. Reprod. 2019, 34, 2201–2207. [Google Scholar]
- Sun, L.; Fang, X.; Chen, Z.; Zhang, H.; Zhang, Z.; Zhou, P.; Xue, T.; Peng, X.; Zhu, Q.; Yin, M.; et al. Compound heterozygous ZP1 mutations cause empty follicle syndrome in infertile sisters. Hum. Mutat. 2019, 40, 2001–2006. [Google Scholar] [CrossRef]
- Chen, T.; Bian, Y.; Liu, X.; Zhao, S.; Wu, K.; Yan, L.; Li, M.; Yang, Z.; Liu, H.; Zhao, H.; et al. A recurrent missense mutation in ZP3 causes empty follicle syndrome and female infertility. Am. J. Hum. Genet. 2017, 101, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, K.; Bai, D.; Yin, J.; Tang, Y.; Chi, F.; Zhang, L.; Wang, Y.; Pan, J.; Liang, S.; et al. Dosage effects of ZP2 and ZP3 heterozygous mutations cause human infertility. Hum. Genet. 2017, 136, 975–985. [Google Scholar] [CrossRef]
- Dai, C.; Hu, L.; Gong, F.; Tan, Y.; Cai, S.; Zhang, S.; Dai, J.; Lu, C.; Chen, J.; Chen, Y.; et al. ZP2 pathogenic variants cause in vitro fertilization failure and female infertility. Genet. Med. 2019, 21, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Pizot, C.; Redin, C.; Piton, A.; Vasli, N.; Stoetzel, C.; Blavier, A.; Laporte, J.; Muller, J. VaRank: A simple and powerful tool for ranking genetic variants. PeerJ. 2015, 3, e796. [Google Scholar] [CrossRef] [PubMed]
- Okutman, O.; Muller, J.; Baert, Y.; Serdarogullari, M.; Gultomruk, M.; Piton, A.; Rombaut, C.; Benkhalifa, M.; Teletin, M.; Skory, V.; et al. Exome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish family. Hum. Mol. Genet. 2015, 24, 5581–5588. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic. Acid. Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Nishimura, K.; Dioguardi, E.; Nishio, S.; Villa, A.; Han, L.; Matsuda, T.; Jovine, L. Molecular basis of egg coat cross-linking sheds light on ZP1-associated female infertility. Nat. Commun. 2019, 10, 3086. [Google Scholar] [CrossRef]
- Rankin, T.; Talbot, P.; Lee, E.; Dean, J. Abnormal zonae pellucidae I mice lacking ZP1 result in early embryonic loss. Development 1999, 126, 3847–3855. [Google Scholar]
- Jimenez-Movilla, M.; Dean, J. ZP2 and ZP3 cytoplasmic tails prevent premature interactions and ensure incorporation into the zona pellucida. J. Cell Sci. 2011, 124, 940–950. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Karyotype | Cycle | Collected oocytes | Results |
|---|---|---|---|---|---|
| VI:2 | 38 | 46,XX | 1-4 | No oocyte OR no MII | No ET, cancelled |
| 5 | 7 oocytes, 2xMII | ET, no pregnancy | |||
| 6 | 10 follicles were aspirated, no oocytes | No ET, cancelled | |||
| 7 | 8 oocytes, no MII | No ET, cancelled | |||
| VI:3 | 36 | 46,XX | 1 | 8 oocytes, no MII | No ET, cancelled |
| VI:4 | 28 | 46,XX | 1 | 11 oocytes, no MII | No ET, cancelled |
| Sample ID | Yield-GB | %Align | Coverage above X25 |
|---|---|---|---|
| VI:2 | 6.125 | 98.85 | 99.6 |
| VI:4 | 5.859 | 98.91 | 99.5 |
| V:3 | 6.913 | 99.6 | 98.88 |
| No | cDNA | aa Change | Zygosity | Phenotype | Ref |
|---|---|---|---|---|---|
| 1 | c.123C>A | p.Tyr41* | Comp Het(1) | No oocyte | [27] |
| 2 | c.170-174del | p.Gly57Aspfs*9 | Comp Het(2) | No oocyte | [28] |
| 3 | c.181C>T | p.Arg61Cys | Comp Het(3) | No oocyte | [26] |
| 4 | c.247T>C | p.Trp83Arg | Het* | Degenerate oocyte | [23] |
| 5 | c.507del | p.His170Ilefs*52 | Hom | No oocyte | [25] |
| 6 | c.508del | p.His170Ile*52 | Comp Het(4) | No oocyte | [27] |
| 7 | c.1014+1G>A | p.? | Hom | No oocyte | [27] |
| 8 | c.1129-1130del | p.Val377Leufs*5 | Hom | No oocyte | [27] |
| 9 | c.1169-1176del | p.Ile390Thrfs*16 | Comp Het(2,3) Hom | No oocyte Zona free oocytes | [26,28] [24] |
| 10 | c.1228C>T | p.Arg410Trp | Hom | Degenerate oocyte | [25] |
| 11 | c.1413G>A | p.Trp471* | Het* | Degenerate oocyte | [23] |
| 12 | c.1430+1G>T | p.? | Comp Het(5) | Zona free oocytes | [25] |
| 13 | c.1510C>T | p.Arg504* | Hom | No oocyte | [27] |
| 14 | c.1573-2A>G | p.? | Comp Het(4) | No oocyte | [27] |
| 15 | c.1663C>T | p.Arg555* | Comp Het(1) | No oocyte | [27] |
| 16 | c.1708G>A | p.Val570Met | Hom | No oocyte or zona free oocytes | [25] |
| 17 | c.1775-8T>C | p.? | Comp Het(5) | Zona free oocytes | [25] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okutman, Ö.; Demirel, C.; Tülek, F.; Pfister, V.; Büyük, U.; Muller, J.; Charlet-Berguerand, N.; Viville, S. Homozygous Splice Site Mutation in ZP1 Causes Familial Oocyte Maturation Defect. Genes 2020, 11, 382. https://doi.org/10.3390/genes11040382
Okutman Ö, Demirel C, Tülek F, Pfister V, Büyük U, Muller J, Charlet-Berguerand N, Viville S. Homozygous Splice Site Mutation in ZP1 Causes Familial Oocyte Maturation Defect. Genes. 2020; 11(4):382. https://doi.org/10.3390/genes11040382
Chicago/Turabian StyleOkutman, Özlem, Cem Demirel, Firat Tülek, Veronique Pfister, Umut Büyük, Jean Muller, Nicolas Charlet-Berguerand, and Stéphane Viville. 2020. "Homozygous Splice Site Mutation in ZP1 Causes Familial Oocyte Maturation Defect" Genes 11, no. 4: 382. https://doi.org/10.3390/genes11040382
APA StyleOkutman, Ö., Demirel, C., Tülek, F., Pfister, V., Büyük, U., Muller, J., Charlet-Berguerand, N., & Viville, S. (2020). Homozygous Splice Site Mutation in ZP1 Causes Familial Oocyte Maturation Defect. Genes, 11(4), 382. https://doi.org/10.3390/genes11040382