Supercoiling, R-Loops, Replication and the Functions of Bacterial Type 1A Topoisomerases

{kind=link}
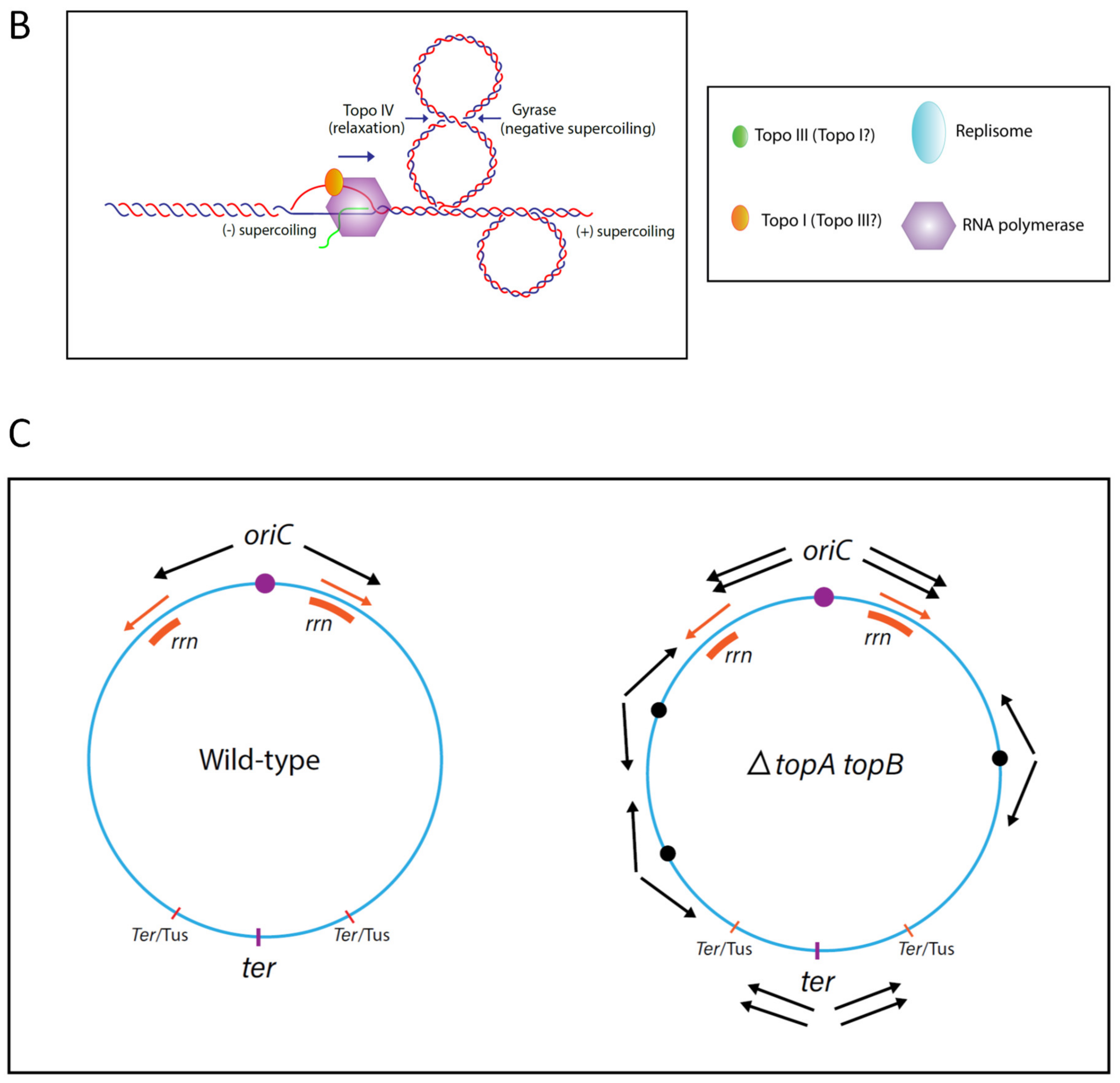{kind=link}
Abstract
1. Introduction
2. Viability of Single topa Null Mutants and the Role of Topo I in Supercoiling Regulation
3. The Role of Topo I in the Regulation of R-loop Formation in E. coli
4. The Main Function of Topo III in E. coli
5. Viability of Double topA topB Null Mutants
6. Topo III and RecQ: Lack of Experimental Evidence for the Presence of a “Toposome” in E. coli
7. Topo IV Overproduction Allows topA topB Null Mutants of E. coli and B. subtilis to be Viable
8. R-loop and RecA in Unregulated Replication in topA topB Null Mutants
9. Topo III: A Specific Role in the Regulation of R-loop Formation or Simply a Back-up for Topo I?
10. Topo I in the Regulation of Replication from oriC
11. A Major Problems of E. coli Cells Lacking Type1A Topos: Over-Replication Leading to Topological Stress and Genomic Instability
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell. Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chan, N.L.; Hsieh, T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Xue, Y.; Lee, S.K.; Martindale, J.L.; Shen, W.; Li, W.; Zou, S.; Ciaramella, M.; Debat, H.; Nadal, M.; et al. RNA topoisomerase is prevalent in all domains of life and associates with polyribosomes in animals. Nucleic Acids Res. 2016, 44, 6335–6349. [Google Scholar] [CrossRef] [PubMed]
- Garnier, F.; Debat, H.; Nadal, M. Type IA DNA Topoisomerases: A Universal Core and Multiple Activities. Methods Mol. Biol. 2018, 1703, 1–20. [Google Scholar] [PubMed]
- Wang, J.C. Interaction between DNA and an Escherichia coli protein omega. J. Mol. Biol. 1971, 55, 523–533. [Google Scholar] [CrossRef]
- Mills, M.; Tse-Dinh, Y.C.; Neuman, K.C. Direct observation of topoisomerase IA gate dynamics. Nat. Struct. Mol. Biol. 2018, 25, 1111–1118. [Google Scholar] [CrossRef]
- DiGate, R.J.; Marians, K.J. Identification of a potent decatenating enzyme from Escherichia coli. J. Biol. Chem. 1988, 263, 13366–13373. [Google Scholar]
- Broccoli, S.; Phoenix, P.; Drolet, M. Isolation of the topB gene encoding DNA topoisomerase III as a multicopy suppressor of topA null mutations in Escherichia coli. Mol. Microbiol. 2000, 35, 58–68. [Google Scholar] [CrossRef]
- Kim, R.A.; Wang, J.C. Identification of the yeast TOP3 gene product as a single strand-specific DNA topoisomerase. J. Biol. Chem. 1992, 267, 17178–17185. [Google Scholar]
- Wilson, T.M.; Chen, A.D.; Hsieh, T. Cloning and characterization of Drosophila topoisomerase IIIbeta. Relaxation of hypernegatively supercoiled DNA. J. Biol. Chem. 2000, 275, 1533–1540. [Google Scholar] [CrossRef]
- Terekhova, K.; Gunn, K.H.; Marko, J.F.; Mondragon, A. Bacterial topoisomerase I and topoisomerase III relax supercoiled DNA via distinct pathways. Nucleic Acids Res. 2012, 40, 10432–10440. [Google Scholar] [CrossRef] [PubMed]
- Terekhova, K.; Marko, J.F.; Mondragon, A. Single-molecule analysis uncovers the difference between the kinetics of DNA decatenation by bacterial topoisomerases I and III. Nucleic Acids Res. 2014, 42, 11657–11667. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Pongpech, P.; DiGate, R.J. Type I topoisomerase activity is required for proper chromosomal segregation in Escherichia coli. Proc. Natl. Acad. Sci. USA 2001, 98, 9766–9771. [Google Scholar] [CrossRef] [PubMed]
- Brochu, J.; Vlachos-Breton, E.; Sutherland, S.; Martel, M.; Drolet, M. Topoisomerases I and III inhibit R-loop formation to prevent unregulated replication in the chromosomal Ter region of Escherichia coli. PLoS Genet. 2018, 14, e1007668. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.R.; Fasshauer, P.; Mroch, P.J.; Ul-Haq, I.; Koo, B.M.; Pohlein, A.; Gross, C.A.; Daniel, R.; Brantl, S.; Stulke, J. Topoisomerase IV can functionally replace all type 1A topoisomerases in Bacillus subtilis. Nucleic Acids Res. 2019, 47, 5231–5242. [Google Scholar] [CrossRef]
- Zechiedrich, E.L.; Khodursky, A.B.; Bachellier, S.; Schneider, R.; Chen, D.; Lilley, D.M.; Cozzarelli, N.R. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000, 275, 8103–8113. [Google Scholar] [CrossRef]
- Nurse, P.; Levine, C.; Hassing, H.; Marians, K.J. Topoisomerase III can serve as the cellular decatenase in Escherichia coli. J. Biol. Chem. 2003, 278, 8653–8660. [Google Scholar] [CrossRef]
- Perez-Cheeks, B.A.; Lee, C.; Hayama, R.; Marians, K.J. A role for topoisomerase III in Escherichia coli chromosome segregation. Mol. Microbiol. 2012, 86, 1007–1022. [Google Scholar] [CrossRef]
- Kogoma, T.; Skarstad, K.; Boye, E.; von Meyenburg, K.; Steen, H.B. RecA protein acts at the initiation of stable DNA replication in rnh mutants of Escherichia coli K-12. J. Bacteriol. 1985, 163, 439–444. [Google Scholar] [CrossRef]
- Dimude, J.U.; Stockum, A.; Midgley-Smith, S.L.; Upton, A.L.; Foster, H.A.; Khan, A.; Saunders, N.J.; Retkute, R.; Rudolph, C.J. The Consequences of Replicating in the Wrong Orientation: Bacterial Chromosome Duplication without an Active Replication Origin. mBio 2015, 6, e01294-15. [Google Scholar] [CrossRef]
- Martel, M.; Balleydier, A.; Sauriol, A.; Drolet, M. Constitutive stable DNA replication in Escherichia coli cells lacking type 1A topoisomerase activity. DNA Repair (Amst) 2015, 35, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Usongo, V.; Drolet, M. Roles of type 1A topoisomerases in genome maintenance in Escherichia coli. PLoS Genet. 2014, 10, e1004543. [Google Scholar] [CrossRef] [PubMed]
- Sternglanz, R.; DiNardo, S.; Voelkel, K.A.; Nishimura, Y.; Hirota, Y.; Becherer, K.; Zumstein, L.; Wang, J.C. Mutations in the gene coding for Escherichia coli DNA topoisomerase I affect transcription and transposition. Proc. Natl. Acad. Sci. USA 1981, 78, 2747–2751. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, S.; Voelkel, K.A.; Sternglanz, R.; Reynolds, A.E.; Wright, A. Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell 1982, 31, 43–51. [Google Scholar] [CrossRef]
- Pruss, G.J.; Manes, S.H.; Drlica, K. Escherichia coli DNA topoisomerase I mutants: Increased supercoiling is corrected by mutations near gyrase genes. Cell 1982, 31, 35–42. [Google Scholar] [CrossRef]
- Raji, A.; Zabel, D.J.; Laufer, C.S.; Depew, R.E. Genetic analysis of mutations that compensate for loss of Escherichia coli DNA topoisomerase I. J. Bacteriol. 1985, 162, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Dorman, C.J.; Lynch, A.S.; Ni Bhriain, N.; Higgins, C.F. DNA supercoiling in Escherichia coli: TopA mutations can be suppressed by DNA amplifications involving the tolC locus. Mol. Microbiol. 1989, 3, 531–540. [Google Scholar] [CrossRef]
- Kato, J.; Nishimura, Y.; Imamura, R.; Niki, H.; Hiraga, S.; Suzuki, H. New topoisomerase essential for chromosome segregation in E. coli. Cell 1990, 63, 393–404. [Google Scholar] [CrossRef]
- Drolet, M.; Phoenix, P.; Menzel, R.; Masse, E.; Liu, L.F.; Crouch, R.J. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. USA 1995, 92, 3526–3530. [Google Scholar] [CrossRef]
- Stupina, V.A.; Wang, J.C. Viability of Escherichia coli topA mutants lacking DNA topoisomerase I. J. Biol. Chem. 2005, 280, 355–360. [Google Scholar] [CrossRef]
- Stockum, A.; Lloyd, R.G.; Rudolph, C.J. On the viability of Escherichia coli cells lacking DNA topoisomerase I. BMC Microbiol. 2012, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Ni Bhriain, N.; Dorman, C.J. Isolation and characterization of a topA mutant of Shigella flexneri. Mol. Microbiol. 1993, 7, 351–358. [Google Scholar] [CrossRef] [PubMed]
- McNairn, E.; Ni Bhriain, N.; Dorman, C.J. Overexpression of the Shigella flexneri genes coding for DNA topoisomerase IV compensates for loss of DNA topoisomerase I: Effect on virulence gene expression. Mol. Microbiol. 1995, 15, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Brauer-Steppkes, T.; Labigne, A.; Cameron, B.; Drlica, K. Topoisomerase I of Helicobacter pylori: Juxtaposition with a flagellin gene (flaB) and functional requirement of a fourth zinc finger motif. Gene 1998, 210, 151–161. [Google Scholar] [CrossRef]
- Ahmed, W.; Menon, S.; Godbole, A.A.; Karthik, P.V.; Nagaraja, V. Conditional silencing of topoisomerase I gene of Mycobacterium tuberculosis validates its essentiality for cell survival. FEMS Microbiol. Lett. 2014, 353, 116–123. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef]
- Wu, H.Y.; Shyy, S.H.; Wang, J.C.; Liu, L.F. Transcription generates positively and negatively supercoiled domains in the template. Cell 1988, 53, 433–440. [Google Scholar] [CrossRef]
- Rovinskiy, N.; Agbleke, A.A.; Chesnokova, O.; Pang, Z.; Higgins, N.P. Rates of gyrase supercoiling and transcription elongation control supercoil density in a bacterial chromosome. PLoS Genet. 2012, 8, e1002845. [Google Scholar] [CrossRef]
- Sutormin, D.; Rubanova, N.; Logacheva, M.; Ghilarov, D.; Severinov, K. Single-nucleotide-resolution mapping of DNA gyrase cleavage sites across the Escherichia coli genome. Nucleic Acids Res. 2019, 47, 1373–1388. [Google Scholar] [CrossRef]
- Cheng, B.; Zhu, C.X.; Ji, C.; Ahumada, A.; Tse-Dinh, Y.C. Direct interaction between Escherichia coli RNA polymerase and the zinc ribbon domains of DNA topoisomerase I. J. Biol. Chem. 2003, 278, 30705–30710. [Google Scholar] [CrossRef]
- Tiwari, P.B.; Chapagain, P.P.; Banda, S.; Darici, Y.; Uren, A.; Tse-Dinh, Y.C. Characterization of molecular interactions between Escherichia coli RNA polymerase and topoisomerase I by molecular simulations. FEBS Lett. 2016, 590, 2844–2851. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Zhou, Q.; Cheng, B.; Zhang, Z.; Joachimiak, A.; Tse-Dinh, Y.C. Structural basis for suppression of hypernegative DNA supercoiling by E. coli topoisomerase I. Nucleic Acids Res. 2015, 43, 11031–11046. [Google Scholar] [CrossRef] [PubMed]
- Rui, S.; Tse-Dinh, Y.C. Topoisomerase function during bacterial responses to environmental challenge. Front. Biosci. 2003, 8, d256–d263. [Google Scholar] [PubMed]
- Banda, S.; Cao, N.; Tse-Dinh, Y.C. Distinct Mechanism Evolved for Mycobacterial RNA Polymerase and Topoisomerase I Protein-Protein Interaction. J. Mol. Biol. 2017, 429, 2931–2942. [Google Scholar] [CrossRef]
- Ahmed, W.; Sala, C.; Hegde, S.R.; Jha, R.K.; Cole, S.T.; Nagaraja, V. Transcription facilitated genome-wide recruitment of topoisomerase I and DNA gyrase. PLoS Genet. 2017, 13, e1006754. [Google Scholar] [CrossRef]
- Hraiky, C.; Raymond, M.A.; Drolet, M. RNase H overproduction corrects a defect at the level of transcription elongation during rRNA synthesis in the absence of DNA topoisomerase I in Escherichia coli. J. Biol. Chem. 2000, 275, 11257–11263. [Google Scholar] [CrossRef]
- Baaklini, I.; Usongo, V.; Nolent, F.; Sanscartier, P.; Hraiky, C.; Drlica, K.; Drolet, M. Hypernegative supercoiling inhibits growth by causing RNA degradation. J. Bacteriol. 2008, 190, 7346–7356. [Google Scholar] [CrossRef]
- Drolet, M. Growth inhibition mediated by excess negative supercoiling: The interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 2006, 59, 723–730. [Google Scholar] [CrossRef]
- Phoenix, P.; Raymond, M.A.; Masse, E.; Drolet, M. Roles of DNA topoisomerases in the regulation of R-loop formation in vitro. J. Biol. Chem. 1997, 272, 1473–1479. [Google Scholar] [CrossRef]
- Drolet, M.; Bi, X.; Liu, L.F. Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J. Biol. Chem. 1994, 269, 2068–2074. [Google Scholar]
- Roy, D.; Zhang, Z.; Lu, Z.; Hsieh, C.L.; Lieber, M.R. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: A nick can serve as a strong R-loop initiation site. Mol. Cell. Biol. 2010, 30, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Stolz, R.; Sulthana, S.; Hartono, S.R.; Malig, M.; Benham, C.J.; Chedin, F. Interplay between DNA sequence and negative superhelicity drives R-loop structures. Proc. Natl. Acad. Sci. USA 2019, 116, 6260–6269. [Google Scholar] [CrossRef] [PubMed]
- Masse, E.; Phoenix, P.; Drolet, M. DNA topoisomerases regulate R-loop formation during transcription of the rrnB operon in Escherichia coli. J. Biol. Chem. 1997, 272, 12816–12823. [Google Scholar] [CrossRef] [PubMed]
- Masse, E.; Drolet, M. Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J. Biol. Chem. 1999, 274, 16659–16664. [Google Scholar] [CrossRef] [PubMed]
- Masse, E.; Drolet, M. R-loop-dependent hypernegative supercoiling in Escherichia coli topA mutants preferentially occurs at low temperatures and correlates with growth inhibition. J. Mol. Biol. 1999, 294, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Yu, K.; Lieber, M.R. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol. Cell. Biol. 2008, 28, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Usongo, V.; Nolent, F.; Sanscartier, P.; Tanguay, C.; Broccoli, S.; Baaklini, I.; Drlica, K.; Drolet, M. Depletion of RNase HI activity in Escherichia coli lacking DNA topoisomerase I leads to defects in DNA supercoiling and segregation. Mol. Microbiol. 2008, 69, 968–981. [Google Scholar]
- Usongo, V.; Martel, M.; Balleydier, A.; Drolet, M. Mutations reducing replication from R-loops suppress the defects of growth, chromosome segregation and DNA supercoiling in cells lacking topoisomerase I and RNase HI activity. DNA Repair (Amst) 2016, 40, 1–17. [Google Scholar] [CrossRef]
- Yang, Z.; Hou, Q.; Cheng, L.; Xu, W.; Hong, Y.; Li, S.; Sun, Q. RNase H1 Cooperates with DNA Gyrases to Restrict R-Loops and Maintain Genome Integrity in Arabidopsis Chloroplasts. Plant. Cell 2017, 29, 2478–2497. [Google Scholar] [CrossRef]
- Lopez, C.R.; Yang, S.; Deibler, R.W.; Ray, S.A.; Pennington, J.M.; Digate, R.J.; Hastings, P.J.; Rosenberg, S.M.; Zechiedrich, E.L. A role for topoisomerase III in a recombination pathway alternative to RuvABC. Mol. Microbiol. 2005, 58, 80–101. [Google Scholar] [CrossRef]
- Hiasa, H.; Marians, K.J. Topoisomerase III, but not topoisomerase I, can support nascent chain elongation during theta-type DNA replication. J. Biol. Chem. 1994, 269, 32655–32659. [Google Scholar] [PubMed]
- Hiasa, H.; Marians, K.J. Two distinct modes of strand unlinking during theta-type DNA replication. J. Biol. Chem. 1996, 271, 21529–21535. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.D.; Crisona, N.J.; Stone, M.D.; Cozzarelli, N.R. Disentangling DNA during replication: A tale of two strands. Philos. Trans. R. Soc. Lond B Biol. Sci. 2004, 359, 39–47. [Google Scholar] [CrossRef] [PubMed]
- DiGate, R.J.; Marians, K.J. Molecular cloning and DNA sequence analysis of Escherichia coli topB, the gene encoding topoisomerase III. J. Biol. Chem. 1989, 264, 17924–17930. [Google Scholar] [PubMed]
- Lee, C.M.; Wang, G.; Pertsinidis, A.; Marians, K.J. Topoisomerase III Acts at the Replication Fork To Remove Precatenanes. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hiasa, H.; Kumar, U.; DiGate, R.J. The traE gene of plasmid RP4 encodes a homologue of Escherichia coli DNA topoisomerase III. J. Biol. Chem. 1997, 272, 19582–19587. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hiasa, H.; DiGate, R. Characterization of a unique type IA topoisomerase in Bacillus cereus. Mol. Microbiol. 2006, 60, 140–151. [Google Scholar] [CrossRef]
- Usongo, V.; Tanguay, C.; Nolent, F.; Bessong, J.E.; Drolet, M. Interplay between type 1A topoisomerases and gyrase in chromosome segregation in Escherichia coli. J. Bacteriol. 2013, 195, 1758–1768. [Google Scholar] [CrossRef]
- Harmon, F.G.; DiGate, R.J.; Kowalczykowski, S.C. RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: A conserved mechanism for control of DNA recombination. Mol. Cell 1999, 3, 611–620. [Google Scholar] [CrossRef]
- Harmon, F.G.; Brockman, J.P.; Kowalczykowski, S.C. RecQ helicase stimulates both DNA catenation and changes in DNA topology by topoisomerase III. J. Biol. Chem. 2003, 278, 42668–42678. [Google Scholar] [CrossRef]
- Gangloff, S.; McDonald, J.P.; Bendixen, C.; Arthur, L.; Rothstein, R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Mol. Cell. Biol. 1994, 14, 8391–8398. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Plank, J.L.; Wu, J.; Hsieh, T.S. Topoisomerase IIIalpha and Bloom’s helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc. Natl. Acad. Sci. USA 2006, 103, 11118–11123. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P.; Plank, J.L.; Bachrati, C.Z.; Hickson, I.D.; Kowalczykowski, S.C. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat. Struct. Mol. Biol. 2010, 17, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Kogoma, T. Stable DNA replication: Interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997, 61, 212–238. [Google Scholar] [CrossRef]
- Drolet, M.; Brochu, J. R-loop-dependent replication and genomic instability in bacteria. DNA Repair (Amst) 2019, 84, 102693. [Google Scholar] [CrossRef]
- Ogawa, T.; Pickett, G.G.; Kogoma, T.; Kornberg, A. RNase H confers specificity in the dnaA-dependent initiation of replication at the unique origin of the Escherichia coli chromosome in vivo and in vitro. Proc. Natl. Acad. Sci. USA 1984, 81, 1040–1044. [Google Scholar] [CrossRef]
- Kaguni, J.M.; Kornberg, A. Topoisomerase I confers specificity in enzymatic replication of the Escherichia coli chromosomal origin. J. Biol. Chem. 1984, 259, 8578–8583. [Google Scholar]
- Michel, B.; Sandler, S.J. Replication Restart in Bacteria. J. Bacteriol. 2017, 199, e00102-17. [Google Scholar] [CrossRef]
- Kasahara, M.; Clikeman, J.A.; Bates, D.B.; Kogoma, T. RecA protein-dependent R-loop formation in vitro. Genes Dev. 2000, 14, 360–365. [Google Scholar]
- Zaitsev, E.N.; Kowalczykowski, S.C. A novel pairing process promoted by Escherichia coli RecA protein: Inverse DNA and RNA strand exchange. Genes Dev. 2000, 14, 740–749. [Google Scholar]
- Maduike, N.Z.; Tehranchi, A.K.; Wang, J.D.; Kreuzer, K.N. Replication of the Escherichia coli chromosome in RNase HI-deficient cells: Multiple initiation regions and fork dynamics. Mol. Microbiol. 2014, 91, 39–56. [Google Scholar] [CrossRef]
- Banda, S.; Tiwari, P.B.; Darici, Y.; Tse-Dinh, Y.C. Investigating direct interaction between Escherichia coli topoisomerase I and RecA. Gene 2016, 585, 65–70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reckinger, A.R.; Jeong, K.S.; Khodursky, A.B.; Hiasa, H. RecA can stimulate the relaxation activity of topoisomerase I: Molecular basis of topoisomerase-mediated genome-wide transcriptional responses in Escherichia coli. Nucleic Acids Res. 2007, 35, 79–86. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tang, S.; Wu, M.K.Y.; Zhang, R.; Hunter, N. Pervasive and essential roles of the Top3-Rmi1 decatenase orchestrate recombination and facilitate chromosome segregation in meiosis. Mol. Cell 2015, 57, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Fasching, C.L.; Cejka, P.; Kowalczykowski, S.C.; Heyer, W.D. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell 2015, 57, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; De Muyt, A.; Lichten, M. Top3-Rmi1 DNA single-strand decatenase is integral to the formation and resolution of meiotic recombination intermediates. Mol. Cell 2015, 57, 583–594. [Google Scholar] [CrossRef]
- Wahba, L.; Gore, S.K.; Koshland, D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife 2013, 2, e00505. [Google Scholar] [CrossRef]
- Zhang, T.; Wallis, M.; Petrovic, V.; Challis, J.; Kalitsis, P.; Hudson, D.F. Loss of TOP3B leads to increased R-loop formation and genome instability. Open Biol. 2019, 9, 190222. [Google Scholar] [CrossRef]
- Leonard, A.C.; Mechali, M. DNA replication origins. Cold Spring Harb. Perspect. Biol. 2013, 5, a010116. [Google Scholar] [CrossRef]
- Bell, S.P.; Kaguni, J.M. Helicase loading at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010124. [Google Scholar] [CrossRef] [PubMed]
- Nollmann, M.; Crisona, N.J.; Arimondo, P.B. Thirty years of Escherichia coli DNA gyrase: From in vivo function to single-molecule mechanism. Biochimie 2007, 89, 490–499. [Google Scholar] [CrossRef]
- Johnsen, L.; Weigel, C.; von Kries, J.; Moller, M.; Skarstad, K. A novel DNA gyrase inhibitor rescues Escherichia coli dnaAcos mutant cells from lethal hyperinitiation. J. Antimicrob. Chemother. 2010, 65, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Sekimizu, K.; Funnell, B.E.; Kornberg, A. Extensive unwinding of the plasmid template during staged enzymatic initiation of DNA replication from the origin of the Escherichia coli chromosome. Cell 1986, 45, 53–64. [Google Scholar] [CrossRef]
- Louarn, J.; Bouche, J.P.; Patte, J.; Louarn, J.M. Genetic inactivation of topoisomerase I suppresses a defect in initiation of chromosome replication in Escherichia coli. Mol. Gen. Genet. 1984, 195, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, J.A.; Sanderlin, A.G.; Laub, M.T. The Stringent Response Inhibits DNA Replication Initiation in E. coli by Modulating Supercoiling of oriC. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Morafraile, E.C.; Hanni, C.; Allen, G.; Zeisner, T.; Clarke, C.; Johnson, M.C.; Santos, M.M.; Carroll, L.; Minchell, N.E.; Baxter, J.; et al. Checkpoint inhibition of origin firing prevents DNA topological stress. Genes Dev. 2019, 33, 1539–1554. [Google Scholar] [CrossRef]
- Minden, J.S.; Marians, K.J. Escherichia coli topoisomerase I can segregate replicating pBR322 daughter DNA molecules in vitro. J. Biol. Chem. 1986, 261, 11906–11917. [Google Scholar]
- Mundbjerg, K.; Jorgensen, S.W.; Fredsoe, J.; Nielsen, I.; Pedersen, J.M.; Bentsen, I.B.; Lisby, M.; Bjergbaek, L.; Andersen, A.H. Top2 and Sgs1-Top3 Act Redundantly to Ensure rDNA Replication Termination. PLoS Genet. 2015, 11, e1005697. [Google Scholar] [CrossRef]
- De Septenville, A.L.; Duigou, S.; Boubakri, H.; Michel, B. Replication fork reversal after replication-transcription collision. PLoS Genet. 2012, 8, e1002622. [Google Scholar] [CrossRef]
- El Sayyed, H.; Le Chat, L.; Lebailly, E.; Vickridge, E.; Pages, C.; Cornet, F.; Cosentino Lagomarsino, M.; Espeli, O. Mapping Topoisomerase IV Binding and Activity Sites on the E. coli Genome. PLoS Genet. 2016, 12, e1006025. [Google Scholar] [CrossRef] [PubMed]
- Crisona, N.J.; Strick, T.R.; Bensimon, D.; Croquette, V.; Cozzarelli, N.R. Preferential relaxation of positively supercoiled DNA by E. coli topoisomerase IV in single-molecule and ensemble measurements. Genes Dev. 2000, 14, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Dorman, C.J. DNA supercoiling and transcription in bacteria: A two-way street. BMC Mol. Cell. Biol. 2019, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Rani, P.; Nagaraja, V. Genome-wide mapping of Topoisomerase I activity sites reveal its role in chromosome segregation. Nucleic Acids Res. 2019, 47, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Bachar, A.; Itzhaki, E.; Gleizer, S.; Shamshoom, M.; Milo, R.; Antonovsky, N. Point mutations in topoisomerase I alter the mutation spectrum in E. coli and impact the emergence of drug resistance genotypes. Nucleic Acids Res. 2020, 48, 761–769. [Google Scholar] [CrossRef]
- Duggin, I.G.; Wake, R.G.; Bell, S.D.; Hill, T.M. The replication fork trap and termination of chromosome replication. Mol. Microbiol. 2008, 70, 1323–1333. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brochu, J.; Breton, É.-V.; Drolet, M. Supercoiling, R-Loops, Replication and the Functions of Bacterial Type 1A Topoisomerases. Genes 2020, 11, 249. https://doi.org/10.3390/genes11030249
Brochu J, Breton É-V, Drolet M. Supercoiling, R-Loops, Replication and the Functions of Bacterial Type 1A Topoisomerases. Genes. 2020; 11(3):249. https://doi.org/10.3390/genes11030249
Chicago/Turabian StyleBrochu, Julien, Émilie-Vlachos Breton, and Marc Drolet. 2020. "Supercoiling, R-Loops, Replication and the Functions of Bacterial Type 1A Topoisomerases" Genes 11, no. 3: 249. https://doi.org/10.3390/genes11030249
APA StyleBrochu, J., Breton, É.-V., & Drolet, M. (2020). Supercoiling, R-Loops, Replication and the Functions of Bacterial Type 1A Topoisomerases. Genes, 11(3), 249. https://doi.org/10.3390/genes11030249