A Network-Based Approach for Identification of Subtype-Specific Master Regulators in Pancreatic Ductal Adenocarcinoma


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Differential Gene and miRNA Expression Analysis
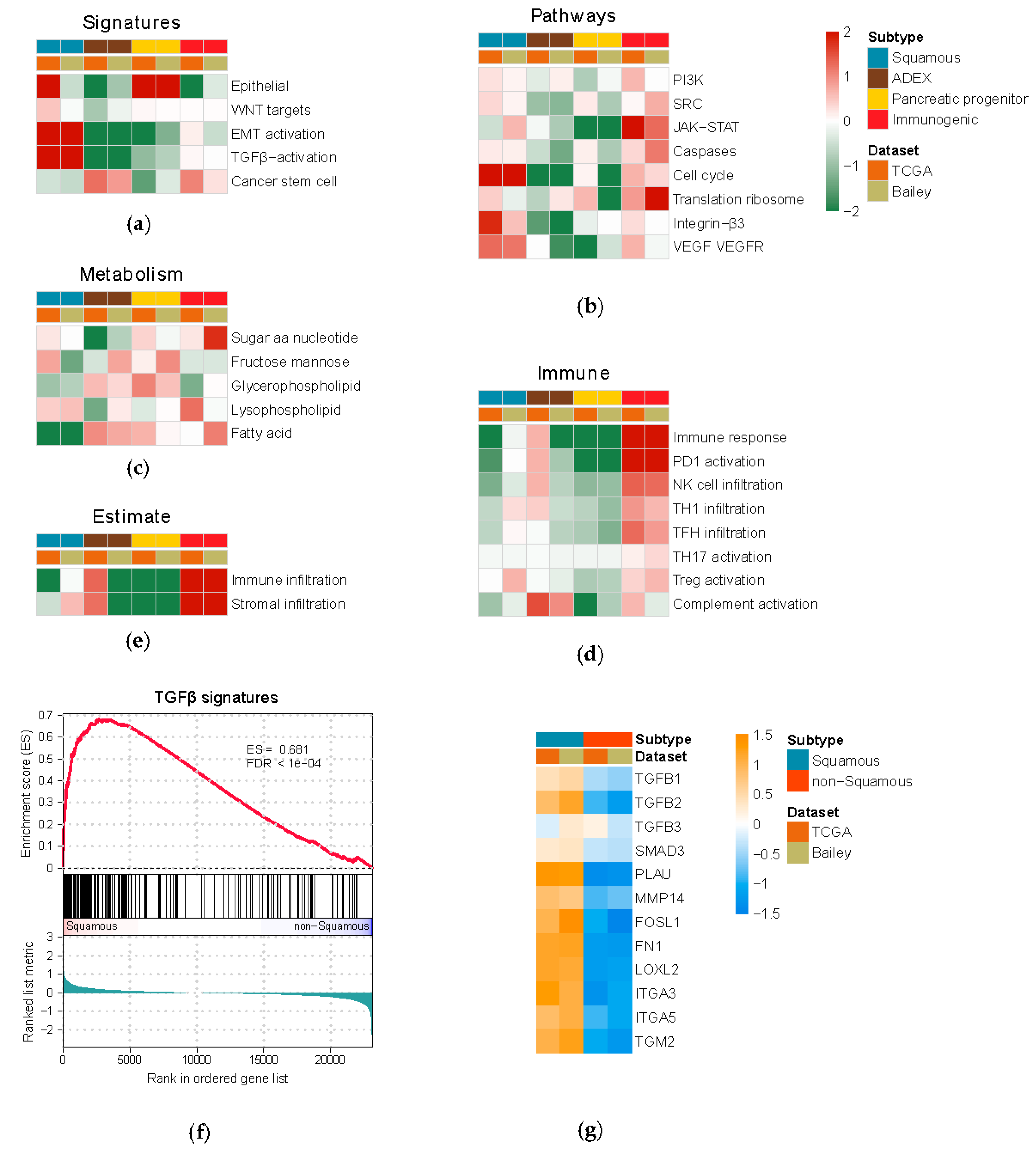
2.3. Gene Set Enrichment Analysis
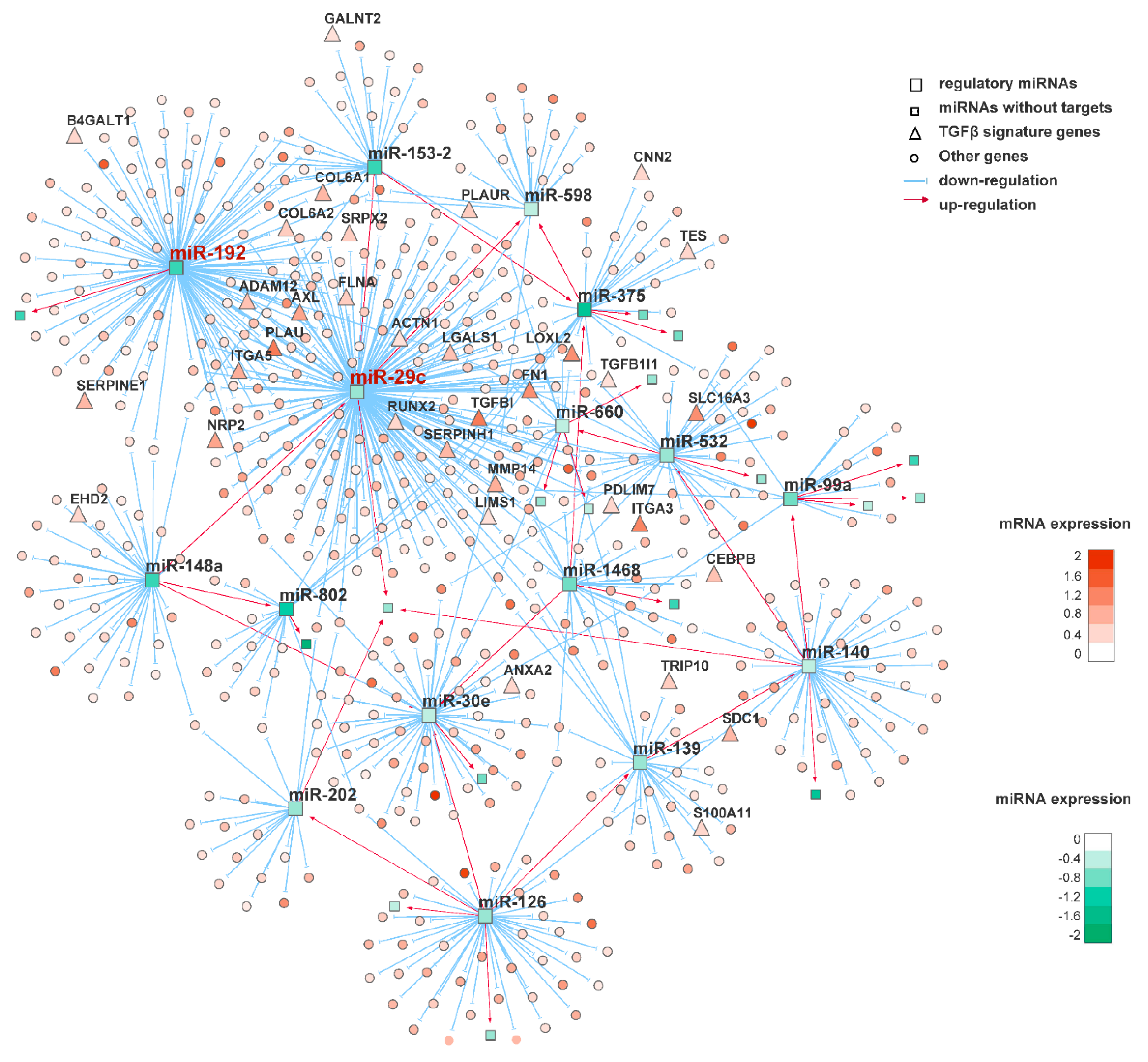
2.4. miRNA-mRNA Regulatory Networks Inference
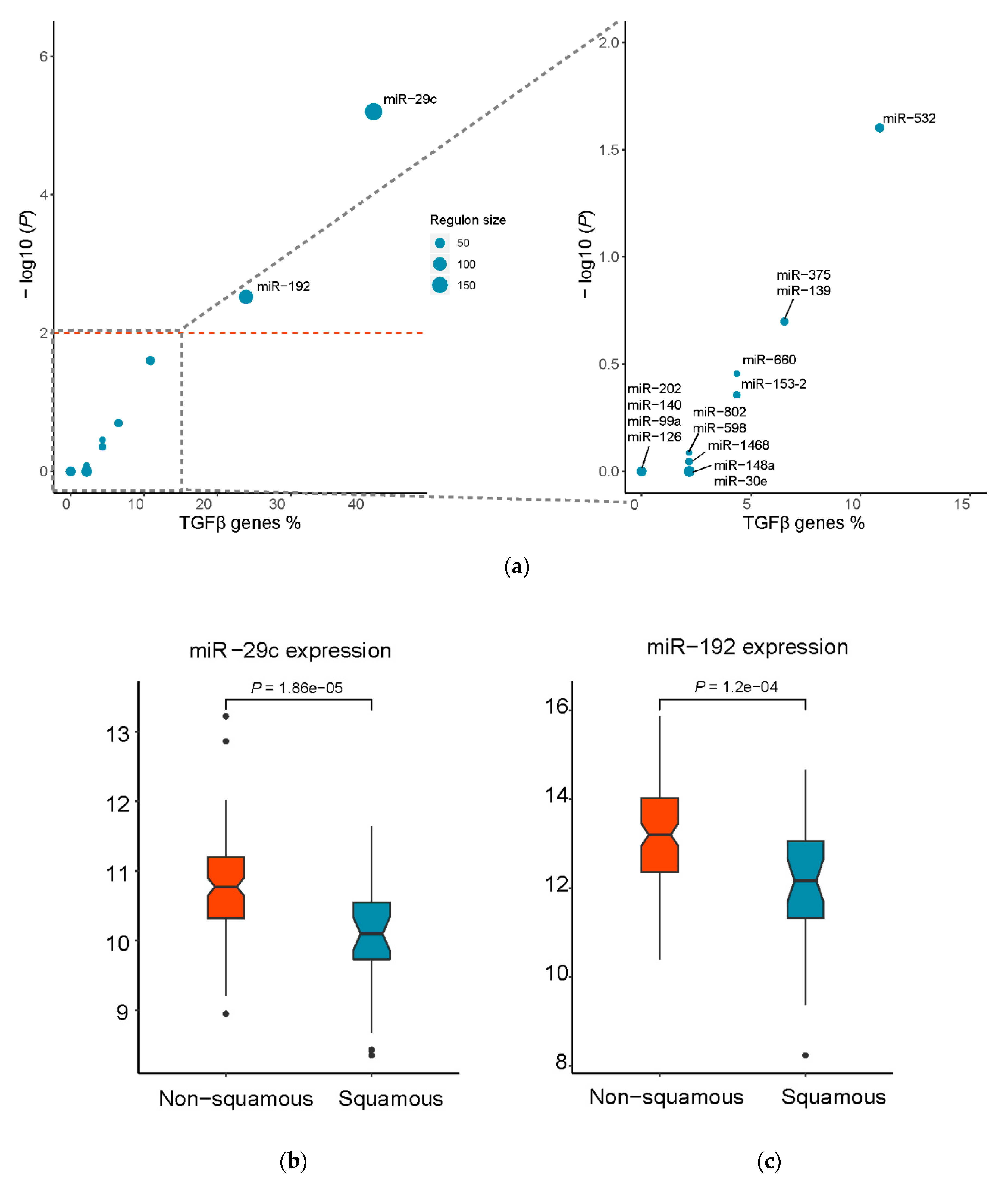
2.5. Master Regulator Analysis
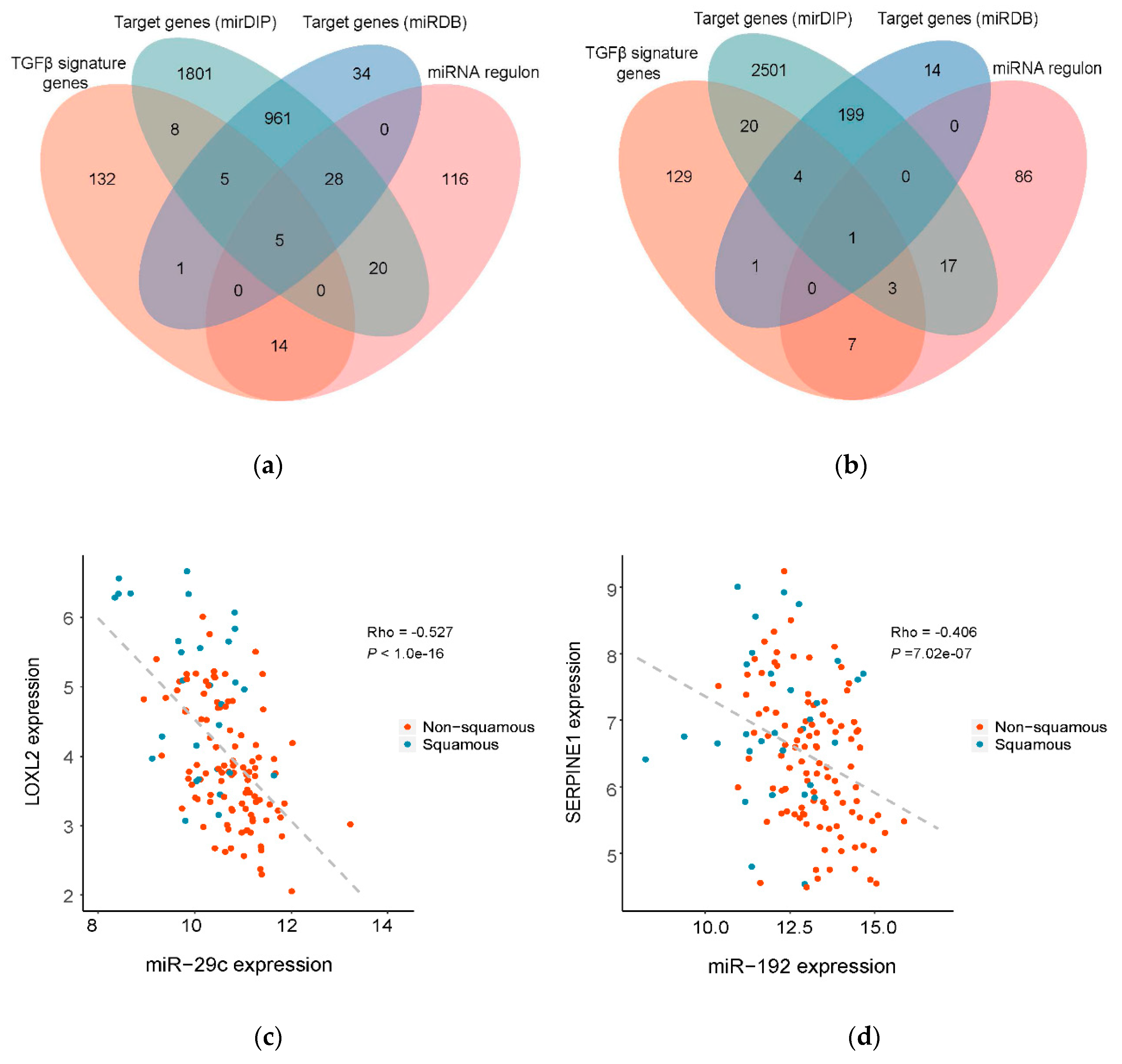
2.6. Prioritization of miRNA Target Genes
2.7. Survival and Drug Sensitivity Analysis
3. Results
3.1. Multi-Dimensional Regulatory Network Inference Based on Integrative Analysis of Gene and miRNA Expression Profiles
3.2. Identification of Putative Master Regulatory miRNAs in the Squamous Subtype of PDAC
3.3. Prioritization of Potential Therapeutic Targets by Identification of the Direct Target Genes of the Master Regulatory miRNAs
3.4. Exploring the Upstream Regulatory Mechanism of the Master Regulatory miRNAs
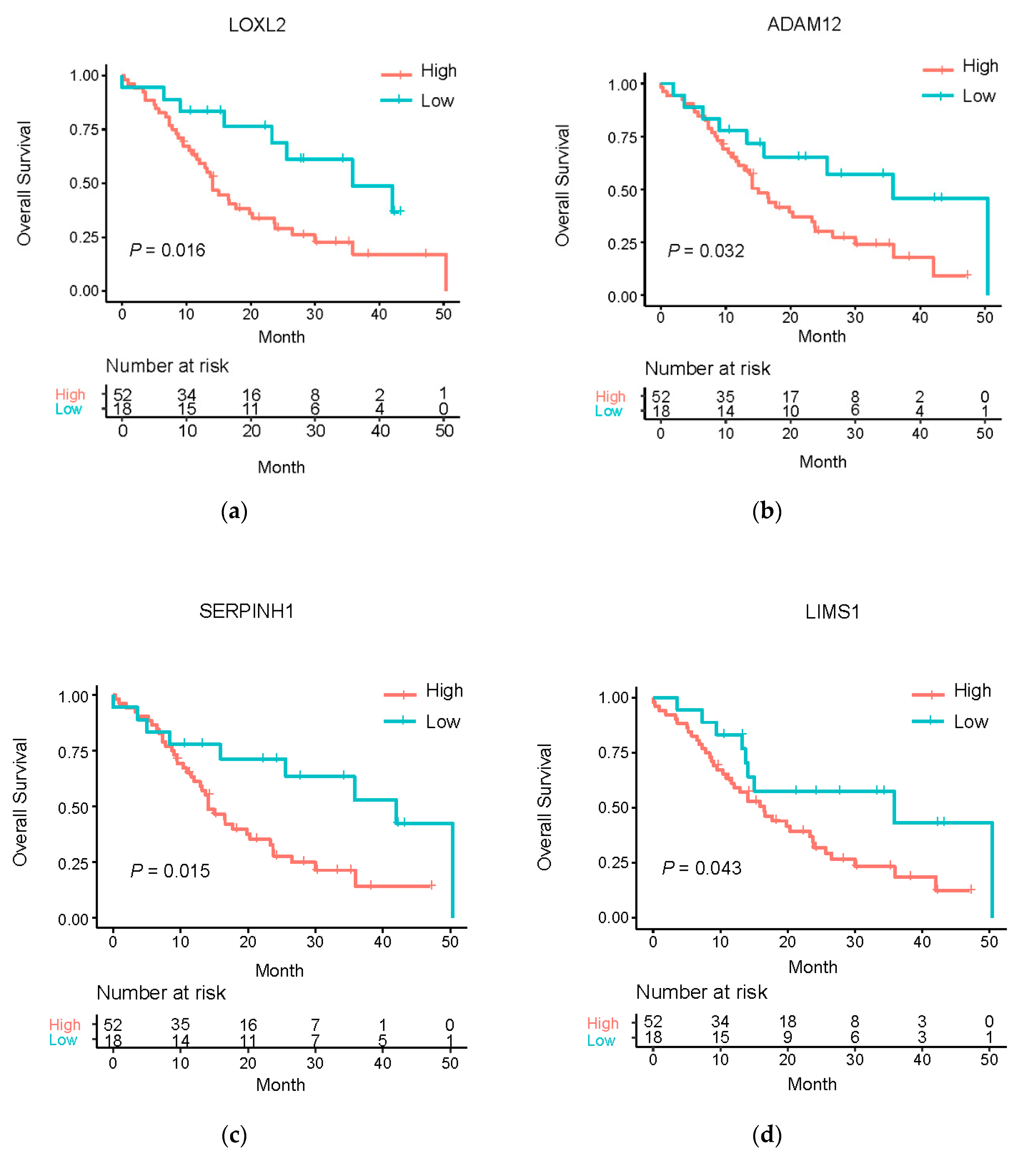
3.5. Clinical Relevance of the Master Regulatory miRNA Target Genes
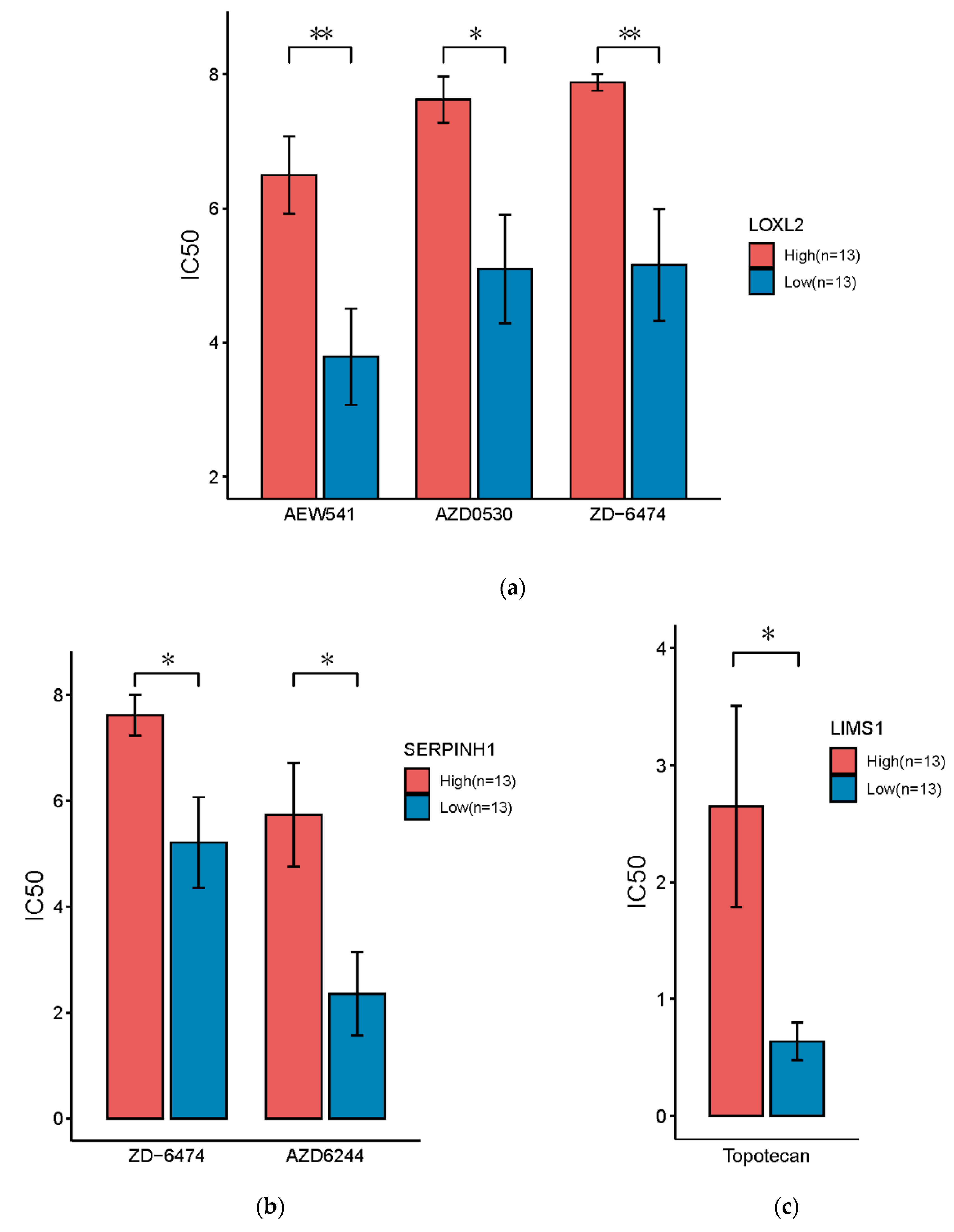
3.6. Drug Sensitivity Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Greither, T.; Grochola, L.F.; Udelnow, A.; Lautenschläger, C.; Würl, P.; Taubert, H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int. J. Cancer 2010, 126, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Siegal, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. Ca. Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J. Borges M. Hruban R.H.; Maitra A. Kinzler K.; Vogelstein B. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Matthaei, H.; Maitra, A.; Dal Molin, M.; Wood, L.D.; Eshleman, J.R.; Goggins, M.; Canto, M.I.; Schulick, R.D.; Edil, B.H. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011, 3, 92ra66. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Kenneth, H.Y.; Yeo, C.J.; Calhoun, E.S. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Bayne, L.J. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol. 2013, 25, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Sjoquist, K.M.; Chin, V.T.; Chantrill, L.A.; O’Connor, C.; Hemmings, C.; Chang, D.K.; Chou, A.; Pajic, M.; Johns, A.L.; Nagrial, A.M. Personalising pancreas cancer treatment: when tissue is the issue. World J. Gastroentero. 2014, 20, 7849–7863. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H. Virtual microdissection identifies distinct tumor-and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Diaz, G.; Zamboni, F.; Tice, A.; Farci, P. Integrated ordination of miRNA and mRNA expression profiles. BMC Genomics 2015, 16, 767. [Google Scholar] [CrossRef]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: molecular basis and therapeutic strategy. Signal Trans. Targ. Therap. 2017, 2, 1–12. [Google Scholar] [CrossRef]
- Fessler, E.; Jansen, M.; Melo, F.D.S.E.; Zhao, L.; Prasetyanti, P.R.; Rodermond, H.; Kandimalla, R.; Linnekamp, J.F.; Franitza, M.; van Hooff, S. A multidimensional network approach reveals microRNAs as determinants of the mesenchymal colorectal cancer subtype. Oncogene 2016, 35, 6026–6037. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, W.; Xu, L.; Yi, T.; Zhao, X.; Wei, Y.; Vermeulen, L.; Goel, A.; Zhou, S.; Wang, X. Integrative network biology analysis identifies miR-508-3p as the determinant for the mesenchymal identity and a strong prognostic biomarker of ovarian cancer. Oncogene 2019, 38, 2305–2319. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: linear models for microarray data. In Bioinformatics and computational biology solutions using R and Bioconductor; Springer Press: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- HTSanalyzeR2. Available online: https://github.com/CityUHK-CompBio/HTSanalyzeR2 (accessed on 1 September 2019).
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Castro, M.A.; de Santiago, I.; Campbell, T.M.; Vaughn, C.; Hickey, T.E.; Ross, E.; Tilley, W.D.; Markowetz, F.; Ponder, B.A.; Meyer, K.B. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 2016, 48, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, M.N.; Castro, M.A.; Wang, X.; De Santiago, I.; O’Reilly, M.; Chin, S.-F.; Rueda, O.M.; Caldas, C.; Ponder, B.A.; Markowetz, F. Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla, F.R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 2006, 7, S7. [Google Scholar] [CrossRef] [PubMed]
- Tokar, T.; Pastrello, C.; Rossos, A.E.; Abovsky, M.; Hauschild, A.-C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1—integrative database of human microRNA target predictions. Nucleic Acids Res. 2017, 46, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2014, 43, 146–152. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Chung, A.C.; Huang, X.R.; Meng, X.; Lan, H.Y. miR-192 mediates TGF-β/Smad3-driven renal fibrosis. J. Am. Soc. Nephr. 2010, 21, 1317–1325. [Google Scholar] [CrossRef]
- Jiang, J.; Yu, C.; Chen, M.; Zhang, H.; Tian, S.; Sun, C. Reduction of miR-29c enhances pancreatic cancer cell migration and stem cell-like phenotype. Oncotarget 2015, 6, 2767–2778. [Google Scholar] [CrossRef]
- Kim, I.-k.; Lee, Y.S.; Kim, H.S.; Dong, S.M.; Park, J.S.; Yoon, D.S. Specific protein 1 (SP1) regulates the epithelial-mesenchymal transition via lysyl oxidase-like 2 (LOXL2) in pancreatic ductal adenocarcinoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Childs, E.J.; Chaffee, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Cote, M.L.; Bondy, M.L.; Hruban, R.H.; Chanock, S.J.; Hoover, R.N. Association of common susceptibility variants of pancreatic cancer in higher-risk patients: a PACGENE study. Cancer Epidemiol. Prevention Biomarkers 2016, 25, 1185–1191. [Google Scholar] [CrossRef]
- Kurahara, H.; Bohl, C.; Natsugoe, S.; Nishizono, Y.; Harihar, S.; Sharma, R.; Iwakuma, T.; Welch, D.R. Suppression of pancreatic cancer growth and metastasis by HMP19 identified through genome-wide shRNA screen. Int. J. Cancer 2016, 139, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, V.; Damhofer, H.; Waasdorp, C.; van Rijssen, L.; van de Vijver, M.; Dijk, F.; Wilmink, H.; Besselink, M.; Busch, O.; Chang, D. ADAM12 is a circulating marker for stromal activation in pancreatic cancer and predicts response to chemotherapy. Oncogenesis 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Li, Y.; Li, Z.; Xu, Y.; Li, N.; Ge, Y.; Dong, J.; Chang, A.; Zhao, T.; Wang, X. LIMS1 Promotes Pancreatic Cancer Cell Survival Under Oxygen-Glucose Deprivation Conditions by Enhancing HIF1A Protein Translation. Clin. Cancer Res. 2019, 25, 4091–4103. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.; Vennin, C.; Conway, J.; Vine, K.L.; Pinese, M.; Cowley, M.J.; Shearer, R.F.; Lucas, M.C.; Herrmann, D.; Allam, A.H. SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene 2017, 36, 4288–4298. [Google Scholar] [CrossRef]
- Botla, S.K.; Savant, S.; Jandaghi, P.; Bauer, A.S.; Mücke, O.; Moskalev, E.A.; Neoptolemos, J.P.; Costello, E.; Greenhalf, W.; Scarpa, A. Early epigenetic downregulation of microRNA-192 expression promotes pancreatic cancer progression. Cancer Res. 2016, 76, 4149–4159. [Google Scholar] [CrossRef]
- Ikenaga, N.; Peng, Z.-W.; Vaid, K.A.; Liu, S.B.; Yoshida, S.; Sverdlov, D.Y.; Mikels-Vigdal, A.; Smith, V.; Schuppan, D.; Popov, Y.V. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017, 66, 1697–1708. [Google Scholar] [CrossRef]
- Pharmaxis releases positive results of Phase 1 clinical trial for LOXL2 inhibitor compound. Available online: https://arixbioscience.com/news/pharmaxis-releases-positive-results-of-phase-1-clinical-trial-for-loxl2-inhibitor-compound (accessed on 1 December 2019).
- Zanders, E.D. Human Drug Targets: A Compendium for Pharmaceutical Discovery; John Wiley & Sons: Chichester, UK, 2015; p. 203. [Google Scholar]
- García-Echeverría, C.; Pearson, M.A.; Marti, A.; Meyer, T.; Mestan, J.; Zimmermann, J.; Gao, J.; Brueggen, J.; Capraro, H.-G.; Cozens, R. In vivo antitumor activity of NVP-AEW541—a novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 2004, 5, 231–239. [Google Scholar]
- Nam, H.-J.; Im, S.-A.; Oh, D.-Y.; Elvin, P.; Kim, H.-P.; Yoon, Y.-K.; Min, A.; Song, S.-H.; Han, S.-W.; Kim, T.-Y. Antitumor activity of saracatinib (AZD0530), a c-Src/Abl kinase inhibitor, alone or in combination with chemotherapeutic agents in gastric cancer. Mol. Cancer Therap. 2013, 12, 16–26. [Google Scholar] [CrossRef]
- Solca, F. Method for treating cancer harboring EGFR mutations. U.S. Patent 8,877,764 B2, 4 November 2014. [Google Scholar]
- Grisham, R.N.; Hyman, D.M.; Iyer, G. Targeted therapies for treatment of recurrent ovarian cancer. Clin. Adv. Hematol. Oncol. 2014, 12, 158–162. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulon | Universe Size | Regulon Size | Total Hits | Expected Hits | Observed Hits | p-value | Adjusted p-value |
|---|---|---|---|---|---|---|---|
| miR-29c | 1530 | 187 | 46 | 5.62 | 19 | 4.00 × 10−07 | 6.30 × 10−06 |
| miR-192 | 1530 | 115 | 46 | 3.46 | 11 | 3.80 × 10−04 | 3.00 × 10−03 |
| Regulator | Target Genes | Log2 FC (Squamous v.s. Non-Squamous) | Adjusted p-Value | Reported Function | References |
|---|---|---|---|---|---|
| miR-29c | COL6A2 | 0.58 | 2.43 × 10−02 | One SNP in COL6A2 (21q22.3) is associated with a higher risk of pancreatic cancer | [42] |
| SERPINH1 | 0.70 | 2.37 × 10−05 | Interacts with HMP19, implicated in PDAC invasion and progression | [43] | |
| LOXL2 | 1.19 | 3.91× 10−06 | Specific protein 1 (SP1) regulates EMT via LOXL2 in PDAC | [41] | |
| ADAM12 | 0.66 | 1.53× 10−02 | A circulating marker for stromal activation in PDAC and can predict response to chemotherapy | [44] | |
| LIMS1 | 0.33 | 7.74× 10−03 | Promotes pancreatic cancer cell survival under specific conditions | [45] | |
| miR-192 | SERPINE1 | 0.78 | 1.17× 10−02 | Mediates tumor invasion and metastasis and acts as a prognostic marker | [46] |
| Gene Symbol | Beta | HR (95% CI for HR) | Wald Test | p-Value |
|---|---|---|---|---|
| LOXL2 | 0.35 | 1.42 (1.06–1.89) | 5.71 | 1.69 × 10−02 |
| ADAM12 | 0.31 | 1.37 (1.00–1.87) | 3.91 | 4.80 × 10−02 |
| COL6A2 | 0.18 | 1.19 (0.84–1.70) | 1.48 | 3.25 × 10−01 |
| SERPINH1 | 0.41 | 1.50 (1.09–2.07) | 6.20 | 1.28 × 10−02 |
| LIMS1 | 0.50 | 1.65 (1.19–2.29) | 9.00 | 2.71 × 10−03 |
| Gene Symbol | Beta | HR (95% CI for HR) | Wald Test | p-Value |
|---|---|---|---|---|
| LOXL2 | 0.27 | 1.31 (1.10–1.58) | 8.67 | 3.24 × 10−03 |
| ADAM12 | 0.22 | 1.25 (1.09–1.43) | 10.80 | 1.02 × 10−03 |
| COL6A2 | 0.37 | 1.45 (1.14–1.84) | 8.98 | 2.73 × 10−03 |
| SERPINH1 | 0.47 | 1.59 (1.19–2.14) | 9.55 | 2.00 × 10−03 |
| LIMS1 | 0.33 | 1.39 (1.01–1.93) | 4.01 | 4.53 × 10−02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhu, L.; Wang, X. A Network-Based Approach for Identification of Subtype-Specific Master Regulators in Pancreatic Ductal Adenocarcinoma. Genes 2020, 11, 155. https://doi.org/10.3390/genes11020155
Zhang Y, Zhu L, Wang X. A Network-Based Approach for Identification of Subtype-Specific Master Regulators in Pancreatic Ductal Adenocarcinoma. Genes. 2020; 11(2):155. https://doi.org/10.3390/genes11020155
Chicago/Turabian StyleZhang, Yuchen, Lina Zhu, and Xin Wang. 2020. "A Network-Based Approach for Identification of Subtype-Specific Master Regulators in Pancreatic Ductal Adenocarcinoma" Genes 11, no. 2: 155. https://doi.org/10.3390/genes11020155
APA StyleZhang, Y., Zhu, L., & Wang, X. (2020). A Network-Based Approach for Identification of Subtype-Specific Master Regulators in Pancreatic Ductal Adenocarcinoma. Genes, 11(2), 155. https://doi.org/10.3390/genes11020155