Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves



 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. RNA Extraction and Sequencing
2.4. Quality Control, Sequence Assembly and Annotation
2.5. Phylogenetic Reconstructions
2.6. Accelerated Evolutionary Rates Analyses
2.7. Identifying Fast Evolving Genes (FEGs) and Positively Selected Genes (PSGs)
2.8. Protein Structure Homology Modelling
3. Results
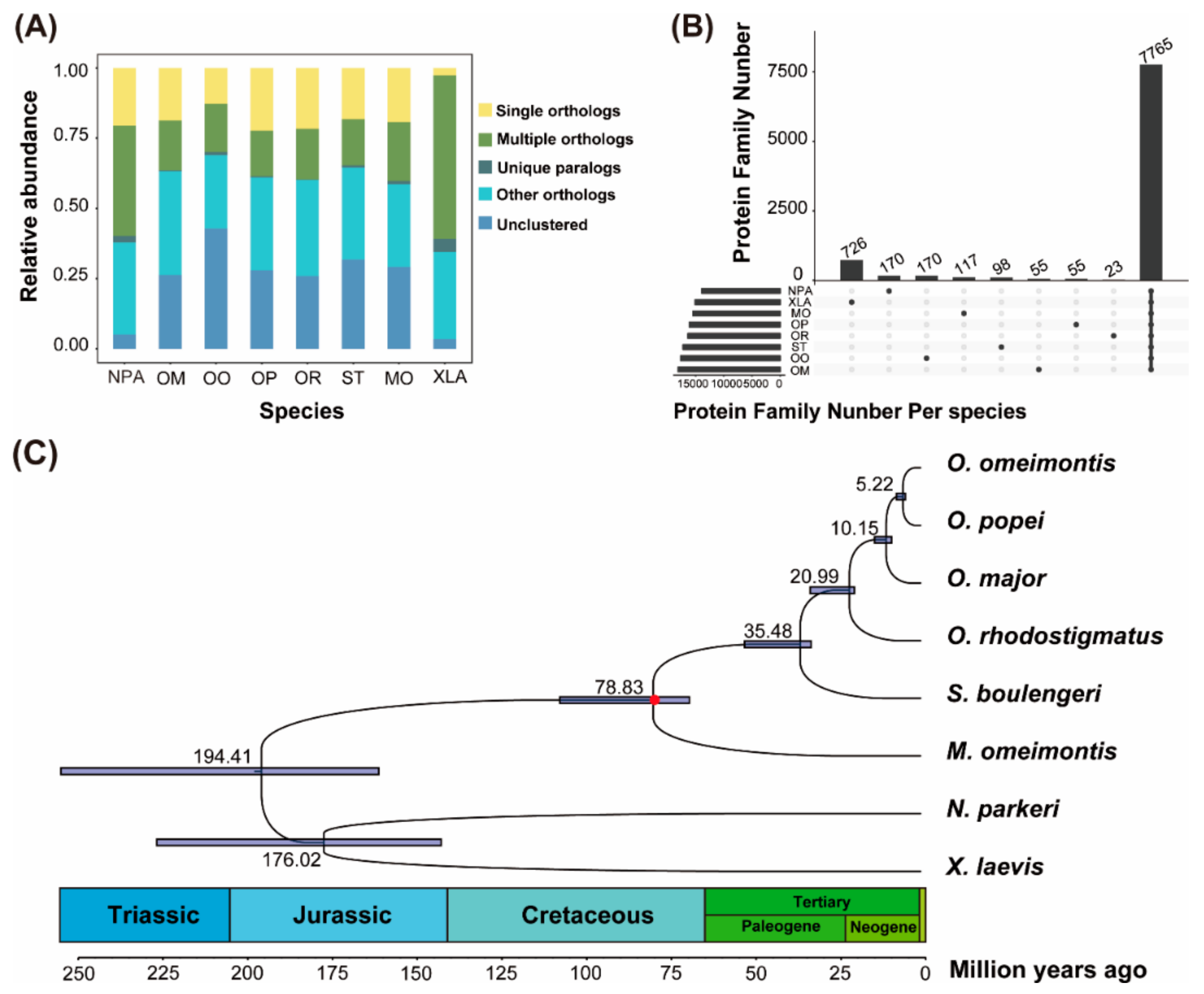
3.1. Gene Homology Analyses
3.2. Phylogenetic Reconstructions
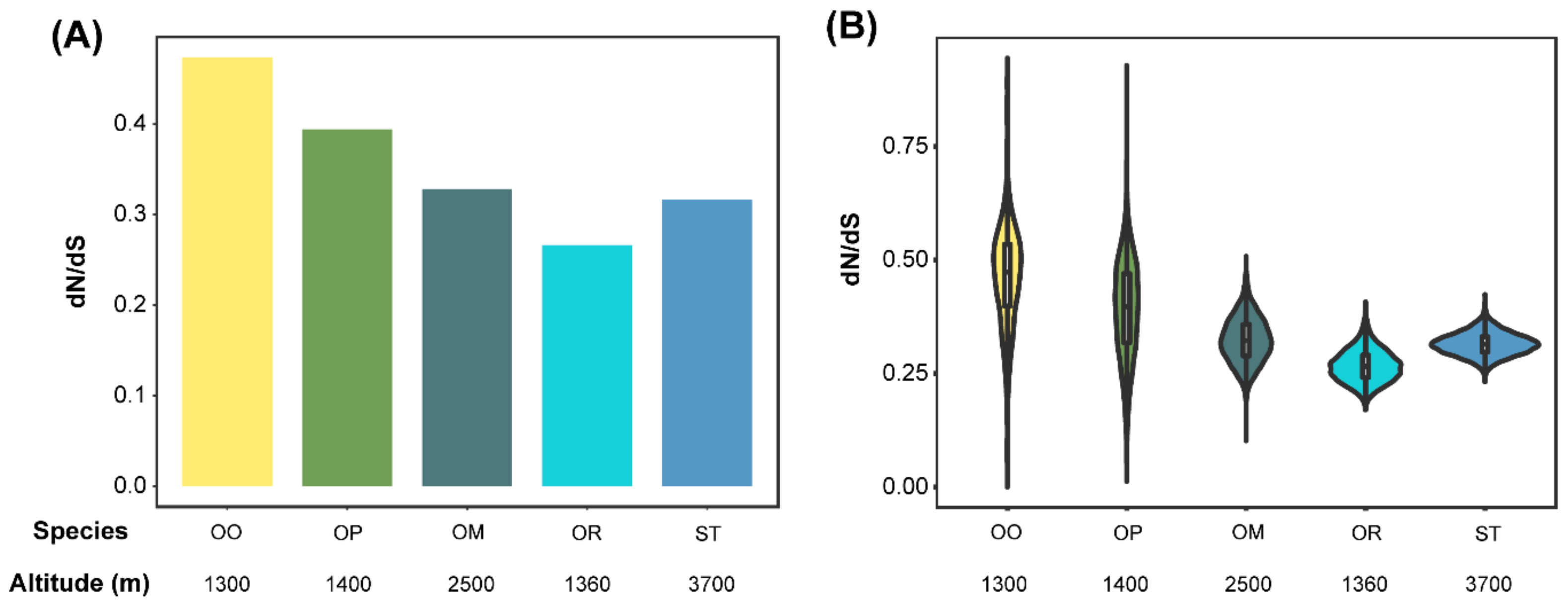
3.3. Accelerated Evolutionary Rates Analyses
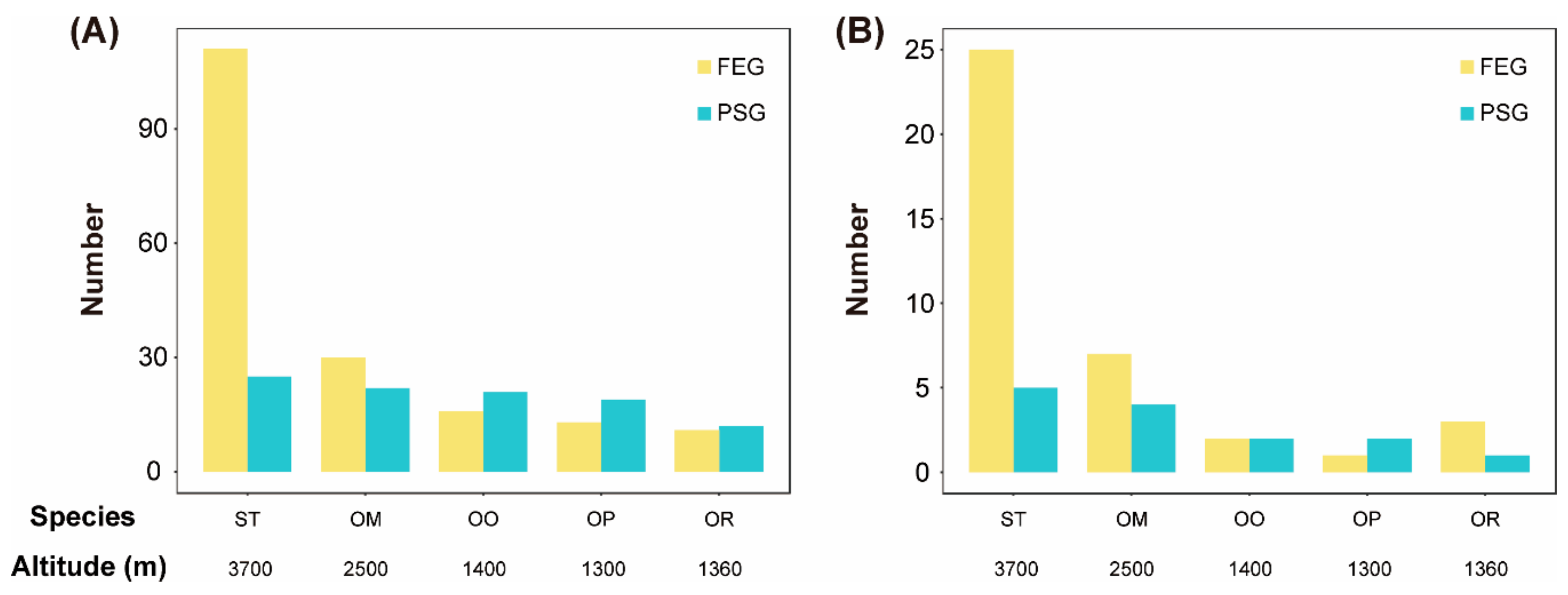
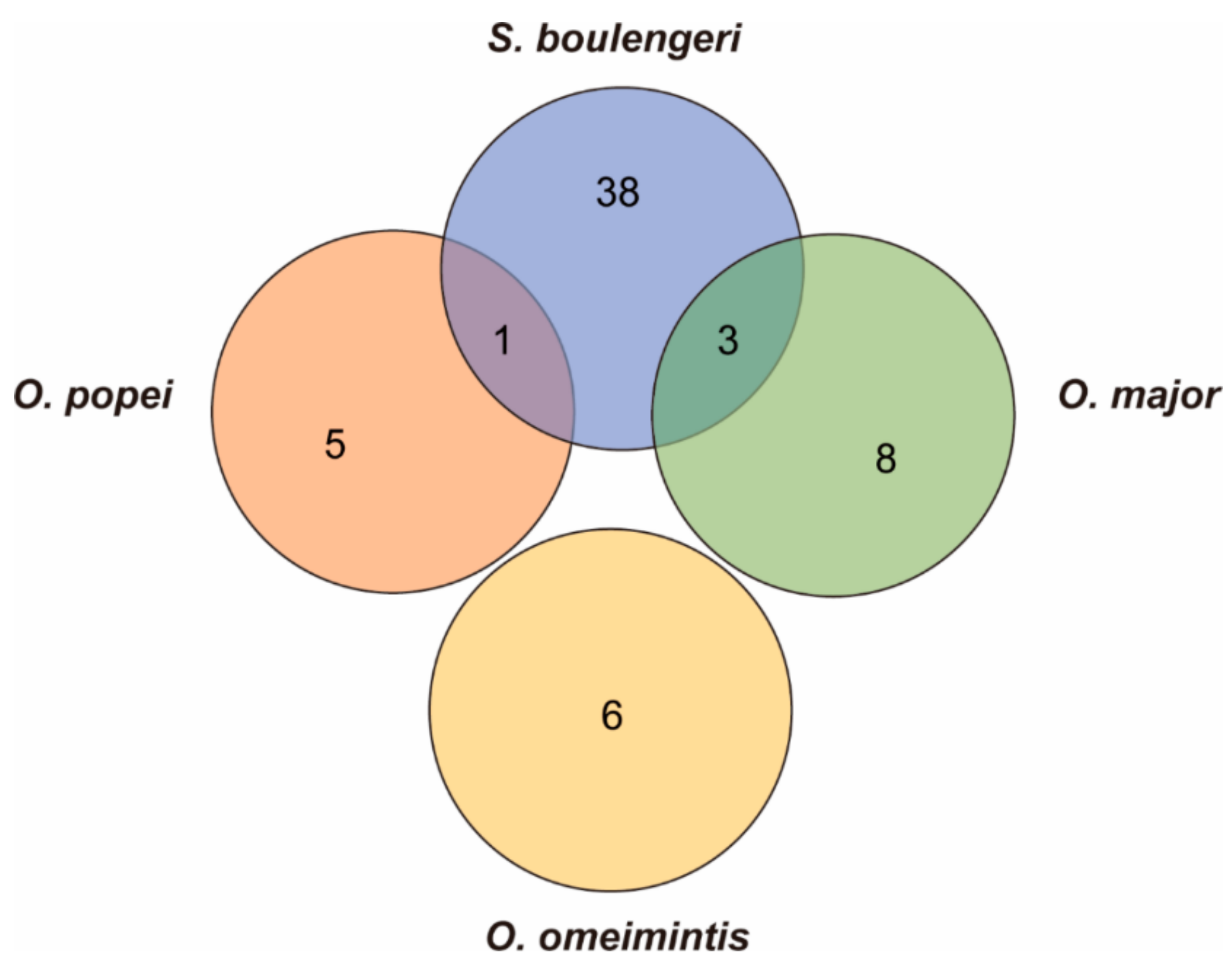
3.4. Fast Evolving Genes (FEGs) and Positively Selected Genes (PSGs)
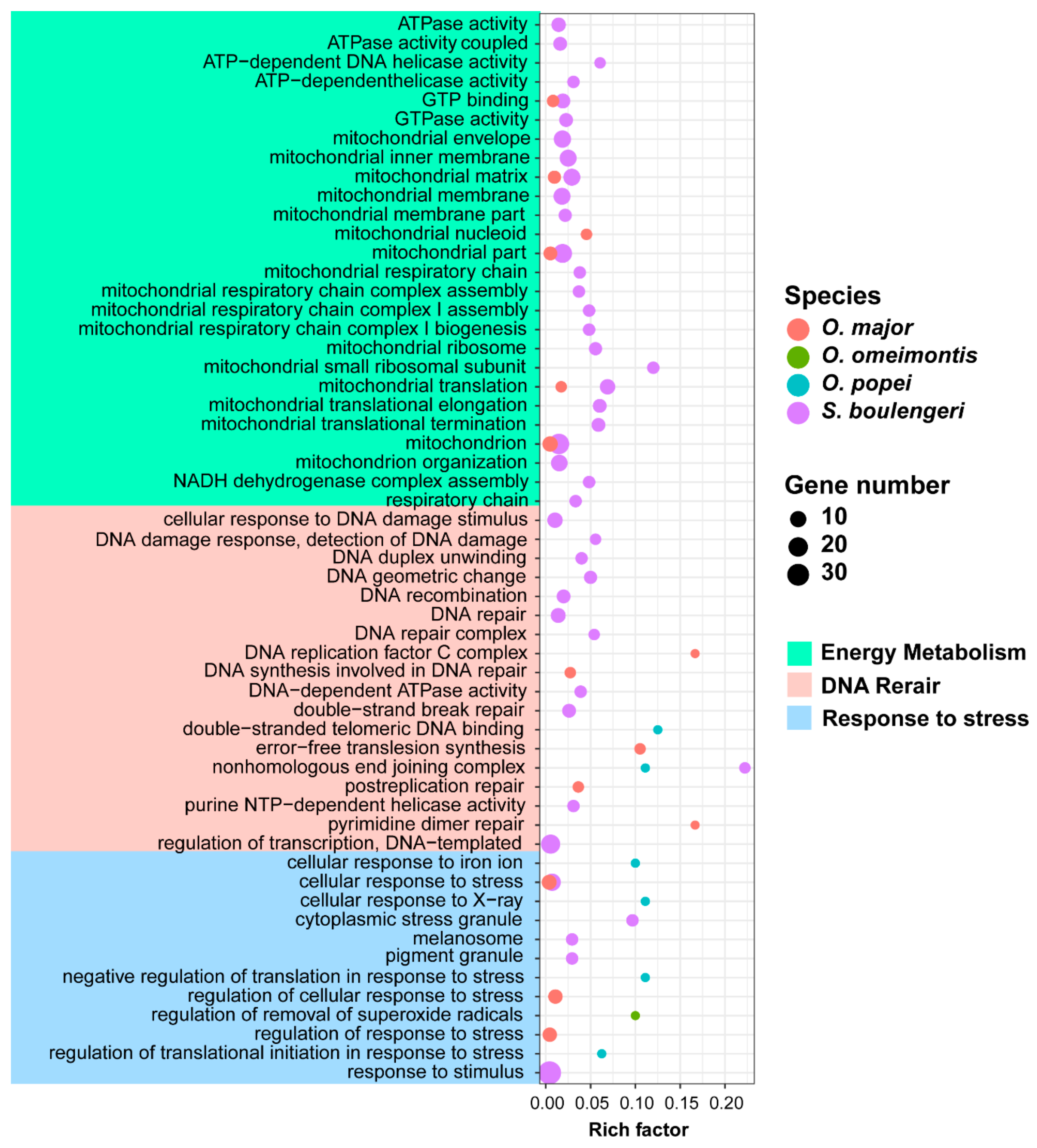
3.5. Candidate Genes for Adapting to High Elevation and Karst Caves
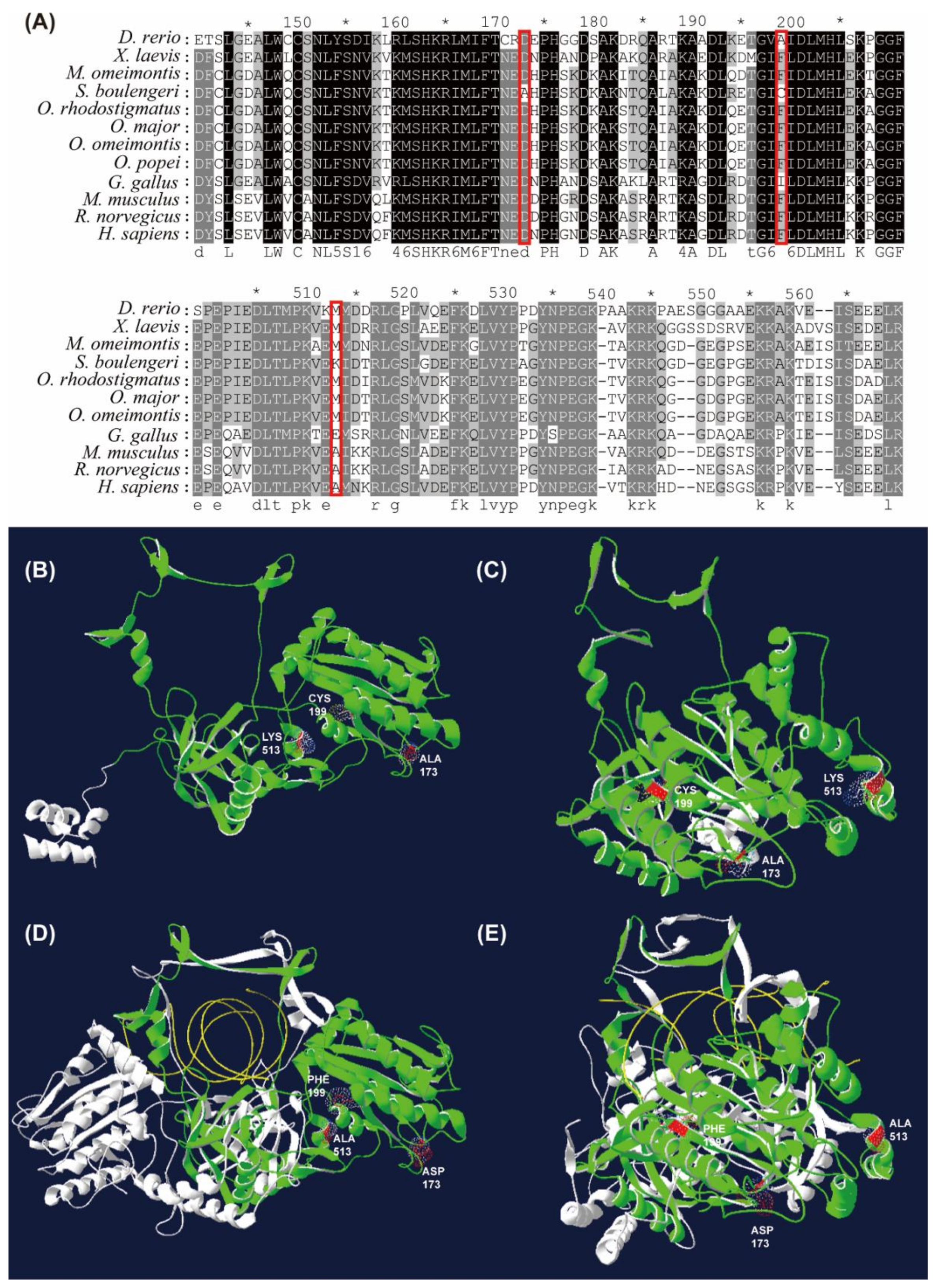
3.6. Amino Acid Substitutions of XRCC6 Gene in S. boulengeri
4. Discussion
4.1. Genetic Signals for High-Elevation Adaptation of Toad Tadpoles on Tibetan Plateau
4.2. Signals of High-Elevation Adaptation Increasing with Elevation in the Toad Species
4.3. Explanations on Genetic Adaptation of the Cave Species O. Rhodostigmatus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rothschild, L.J.; Mancinelli, R.L.J.N. Life in extreme environments. Nature 2001, 409, 1092. [Google Scholar] [CrossRef] [PubMed]
- Taute, K.M.; Gude, S.; Nghe, P.; Tans, S.J. Evolutionary constraints in variable environments, from proteins to networks. Trends Genet. 2014, 30, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kenkel, C.D.; Matz, M.V. Gene expression plasticity as a mechanism of coral adaptation to a variable environment. Nat. Ecol. Evol. 2016, 1, 0014. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-H.; Chang, Y.-T. On Tentative Scheme for Dividing Zoogeographical Regions of China. Acta Geogr. Sin. 1956, 23, 93–109. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Zhang, Y.; Lee, W.-H.; Zhang, Y. Rich diversity and potency of skin antioxidant peptides revealed a novel molecular basis for high-altitude adaptation of amphibians. Sci. Rep. 2016, 6, 19866. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F.; Scott, G.R.; Cheviron, Z.A. Phenotypic plasticity and genetic adaptation to high-altitude hypoxia in vertebrates. J. Exp. Biol. 2010, 213, 4125–4136. [Google Scholar] [CrossRef]
- Simonson, T.S.; Yang, Y.Z.; Huff, C.D.; Yun, H.X.; Qin, G.; Witherspoon, D.J.; Bai, Z.Z.; Lorenzo, F.R.; Xing, J.C.; Jorde, L.B.; et al. Genetic Evidence for High-Altitude Adaptation in Tibet. Science 2010, 329, 72–75. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.J.; Ma, T.; Qian, W.B.; Wang, J.Y.; Ye, Z.Q.; Cao, C.C.; Hu, Q.J.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nature Genet. 2012, 44, 946. [Google Scholar] [CrossRef]
- Ge, R.-L.; Cai, Q.; Shen, Y.-Y.; San, A.; Ma, L.; Zhang, Y.; Yi, X.; Chen, Y.; Yang, L.; Huang, Y.; et al. Draft genome sequence of the Tibetan antelope. Nature Commun. 2013, 4. [Google Scholar] [CrossRef]
- Qu, Y.; Zhao, H.; Han, N.; Zhou, G.; Song, G.; Gao, B.; Tian, S.; Zhang, J.; Zhang, R.; Meng, X.; et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nature Commun. 2013, 4, 1858. [Google Scholar] [CrossRef]
- Hao, Y.; Xiong, Y.; Cheng, Y.; Song, G.; Jia, C.; Qu, Y.; Lei, F. Comparative transcriptomics of 3 high-altitude passerine birds and their low-altitude relatives. Proc. Natl. Acad. Sci. USA 2019, 116, 11851–11856. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-B.; Fu, T.-T.; Jin, J.-Q.; Murphy, R.W.; Hillis, D.M.; Zhang, Y.-P.; Che, J. Species groups distributed across elevational gradients reveal convergent and continuous genetic adaptation to high elevations. Proc. Natl. Acad. Sci. USA 2018, 115, E10634–E10641. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-D.; Zhang, B.-L.; Zhou, W.-W.; Li, Y.-X.; Jin, J.-Q.; Shao, Y.; Yang, H.-c.; Liu, Y.-H.; Yan, F.; Chen, H.-M.; et al. Selection and environmental adaptation along a path to speciation in the Tibetan frog Nanorana parkeri. Proc. Natl. Acad. Sci. USA 2018, 115, E5056–E5065. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Fei, T.; Zhang, C.; Zhao, K. Comprehensive transcriptomic analysis of Tibetan Schizothoracinae fish Gymnocypris przewalskii reveals how it adapts to a high altitude aquatic. BMC Evol. Biol. 2017, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Ma, X.; Niu, J.; Zou, M. Genome-wide identification of genes probably relevant to the adaptation of schizothoracins (Teleostei: Cypriniformes) to the uplift of the Qinghai-Tibet Plateau. BMC Genom. 2017, 18, 310. [Google Scholar] [CrossRef] [PubMed]
- Poulson, T.L.; White, W.B. Cave environment. Science 1969, 165, 971–981. [Google Scholar] [CrossRef]
- Romero, A.; Zhao, Y.; Chen, X. The Hypogean fishes of China. In Chinese Fishes; Noakes, D.L.G., Romero, A., Zhao, Y., Zhou, Y., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2010; pp. 211–278. [Google Scholar]
- Jeffery, W.R. Regressive Evolution in Astyanax Cavefish. Annu. Rev. Genet. 2009, 43, 25–47. [Google Scholar] [CrossRef]
- Rohner, N.; Jarosz, D.F.; Kowalko, J.E.; Yoshizawa, M.; Jeffery, W.R.; Borowsky, R.L.; Lindquist, S.; Tabin, C.J. Cryptic Variation in Morphological Evolution: HSP90 as a Capacitor for Loss of Eyes in Cavefish. Science 2013, 342, 1372–1375. [Google Scholar] [CrossRef]
- McGaugh, S.E.; Gross, J.B.; Aken, B.; Blin, M.; Borowsky, R.; Chalopin, D.; Hinaux, H.; Jeffery, W.R.; Keene, A.; Ma, L.; et al. The cavefish genome reveals candidate genes for eye loss. Nat. Commun. 2014, 5, 5307. [Google Scholar] [CrossRef]
- Yang, J.; Chen, X.; Bai, J.; Fang, D.; Qiu, Y.; Jiang, W.; Yuan, H.; Bian, C.; Lu, J.; He, S.; et al. The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 2016, 14, 1. [Google Scholar] [CrossRef]
- Krishnan, J.; Rohner, N. Cavefish and the basis for eye loss. Philos. Trans. R. Soc. B 2017, 372, 20150487. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.Z.; Lathrop, A.; Murphy, R.W. Phylogeny of genus Scutiger (Amphibia: Megophryidae): A re-evaluation. Asiat. Herpetol. Res. 1997, 7, 32–37. [Google Scholar]
- Dubois, A. Notes sur la systématique et la répartition des Amphibiens Anoures de Chine et des régions avoisinantes. IV. Classification générique et subgénérique des Pelobatidae Megophryinae. Publications de la Société Linnéenne de Lyon 1980, 49, 469–482. [Google Scholar] [CrossRef]
- LIu, J. Ontogenesis and primary ecological study of Oreolalax rhodostigmatus. Bull. Biol. 2010, 45, 50–52. [Google Scholar]
- Shen, Y.H.; Gu, Q.; Gu, Z.-R.; Mao, H.Y.O. rhodostigmatus in the North-western Hunan Province: The Cave Life and the Characteristics of the Growth and Development of Its Tadpoles. Life Sci. Res. 2014, 18, 494–499+501. [Google Scholar]
- Alexander Pyron, R.; Wiens, J.J. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol. Phylogenetics Evol. 2011, 61, 543–583. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 26 June 2017).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLOS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of Life Reveals Clock—Like Speciation and Diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Codon-Substitution Models for Detecting Molecular Adaptation at Individual Sites Along Specific Lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xie, F.; Jiang, J. Histological Observation of Skin in Four Species in the Genus Scutiger. Sichuan J. Zool. 2011, 30, 214. [Google Scholar]
- Gronskov, K.; Dooley, C.M.; Ostergaard, E.; Kelsh, R.N.; Hansen, L.; Levesque, M.P.; Vilhelmsen, K.; Mollgard, K.; Stemple, D.L.; Rosenberg, T. Mutations in C10orf11, a Melanocyte-Differentiation Gene, Cause Autosomal-Recessive Albinism. Am. J. Hum. Genet. 2013, 92, 415–421. [Google Scholar] [CrossRef]
- Svobodova, A.R.; Galandakova, A.; Sianska, J.; Dolezal, D.; Lichnovska, R.; Ulrichova, J.; Vostalova, J. DNA damage after acute exposure of mice skin to physiological doses of UVB and UVA light. Arch. Dermatol. Res. 2012, 304, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Mitsuhashi, N.; Miki, T.; Senbongi, H.; Yokoi, N.; Yano, H.; Miyazaki, M.; Nakajima, N.; Iwanaga, T.; Yokoyama, Y.; Shibata, T.; et al. MTABC3, a novel mitochondrial ATP-binding cassette protein involved in iron homeostasis. J. Biol. Chem. 2000, 275, 17536–17540. [Google Scholar] [CrossRef]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Huettemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef]
- Bickler, P.E.; Buck, L.T. Hypoxia tolerance in reptiles, amphibians, and fishes: Life with variable oxygen availability. In Annual Review of Physiology; Annual Reviews: Palo Alto, CA, USA, 2007; Volume 69, pp. 145–170. [Google Scholar]
- Wenz, L.S.; Opalinski, L.; Wiedemann, N.; Becker, T. Cooperation of protein machineries in mitochondrial protein sorting. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 1119–1129. [Google Scholar] [CrossRef]
- Johnston, I.G.; Williams, B.P. Evolutionary Inference across Eukaryotes Identifies Specific Pressures Favoring Mitochondrial Gene Retention. Cell Syst. 2016, 2, 101–111. [Google Scholar] [CrossRef]
- Katusic, Z.S.; Stelter, A.; Milstien, S. Cytokines stimulate GTP cyclohydrolase I gene expression in cultured human umbilical vein endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 27–32. [Google Scholar] [CrossRef]
- Cai, S.; Alp, N.J.; McDonald, D.; Smith, I.; Kay, J.; Canevari, L.; Heales, S.; Channon, K.M. GTP cyclohydrolase I gene transfer augments intracellular tetrahydrobiopterin in human endothelial cells: Effects on nitric oxide synthase activity, protein levels and dimerisation. Cardiovasc. Res. 2002, 55, 838–849. [Google Scholar] [CrossRef][Green Version]
- Smirnova, E.V.; Rakitina, T.V.; Bogatova, O.V.; Ivanova, D.L.; Vorobyeva, E.E.; Lipkin, A.V.; Kostanyan, I.A.; Lipkin, V.M. Novel Protein Haponin Regulates Cellular Response to Oxidative Stress. Dokl. Biochem. Biophys. 2011, 440, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, R.; Palumbo, V.; Di Savino, A.; Velasco, S.; Sbroggio, M.; Sportoletti, P.; Micale, L.; Turco, E.; Silengo, L.; Palumbo, G.; et al. Morgana/chp-1, a ROCK Inhibitor Involved in Centrosome Duplication and Tumorigenesis. Dev. Cell 2010, 18, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Luche, R.; Wei, B.; Gordon, M.L.; Diltz, C.D.; Tonks, N.K. Activation of the Jnk signaling pathway by a dual-specificity phosphatase, JSP-1. Proc. Natl. Acad. Sci. USA 2001, 98, 13613–13618. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Han, J.; Tang, X.; Ma, M.; Wang, K.; Zhang, X.; Ren, Q.; Chen, Q.; Qiu, Q. Comparative transcriptomic analysis revealed adaptation mechanism of Phrynocephalus erythrurus, the highest altitude Lizard living in the Qinghai-Tibet Plateau. BMC Evol. Biol. 2015, 15, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liu, L.; Wang, X.; Gao, X.; Jiang, J.; Wang, B. Transcriptomics reveals the molecular processes of light-induced rapid darkening of the non-obligate cave dweller Oreolalax rhodostigmatus (Megophryidae, Anura) and their genetic basis of pigmentation strategy. BMC Genom. 2018, 19, 422. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | FEGs | PSGs |
|---|---|---|
| S. boulengeri | GFM1, MTIF2, ERAL1, GFM2, MRPL37, MRPS35, MRPS7, MRPS28, OXA1, C7orf55, NDUFA9, NDUFA12, SLC25A32, SLC25A10, RECQL, XRCC6, UBE2M, NSMCE1, RFC1, DDX1, CLDN3, SLC35C2, CHCHD2, ABCB6, CLPB, NPTX1, CHRND, SIGIRR, FAM58A, PMF1, AVEN, E2F6, C10orf11, VPS39 | PDE12, ERAL1, NDUFA9, RECQL, AMN1, AVEN, CAPG, ARMC5, FAM58A, LRMDA, PSMB8, UBE2M, MRPS35 |
| O. major | MTPAP, MTIF2, SCO1, ERAL1, POLH, RFC1, MRGBP, RAB8B, DUSP22 | SCO1, ERAL1, RFC1, MRGBP, HSF2, DUSP22, IDS |
| O. popei | XRCC6, PYROXD1, TFR2, LACTB2, TECTA | ACP7, TAMM41, XRCC6, PYROXD1, TFR2, LACTB2, PINX1 |
| O. omeimontis | PYROXD2, TSG101, EIF1AD, GCH1 | MTG1, CRLS1, GCH1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, L.; Zhu, W.; Shi, S.; Zhang, M.; Jiang, J.; Li, C.; Xie, F.; Wang, B. Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves. Genes 2020, 11, 123. https://doi.org/10.3390/genes11020123
Chang L, Zhu W, Shi S, Zhang M, Jiang J, Li C, Xie F, Wang B. Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves. Genes. 2020; 11(2):123. https://doi.org/10.3390/genes11020123
Chicago/Turabian StyleChang, Liming, Wei Zhu, Shengchao Shi, Meihua Zhang, Jianping Jiang, Cheng Li, Feng Xie, and Bin Wang. 2020. "Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves" Genes 11, no. 2: 123. https://doi.org/10.3390/genes11020123
APA StyleChang, L., Zhu, W., Shi, S., Zhang, M., Jiang, J., Li, C., Xie, F., & Wang, B. (2020). Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves. Genes, 11(2), 123. https://doi.org/10.3390/genes11020123