Identification of Novel CDH23 Variants Causing Moderate to Profound Progressive Nonsyndromic Hearing Loss

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects and Ethical Considerations
2.2. Clinical Evaluation of Subjects
2.3. DNA Extraction and Whole-Genome SNP Genotyping Using AxiomTM 6.0 Array
2.4. Whole Exome Sequencing (WES), Data Processing, and Variant Analysis
2.5. Sanger Validation and Segregation Analysis
2.6. Pathogenicity Computation and In Silico Modeling
3. Results
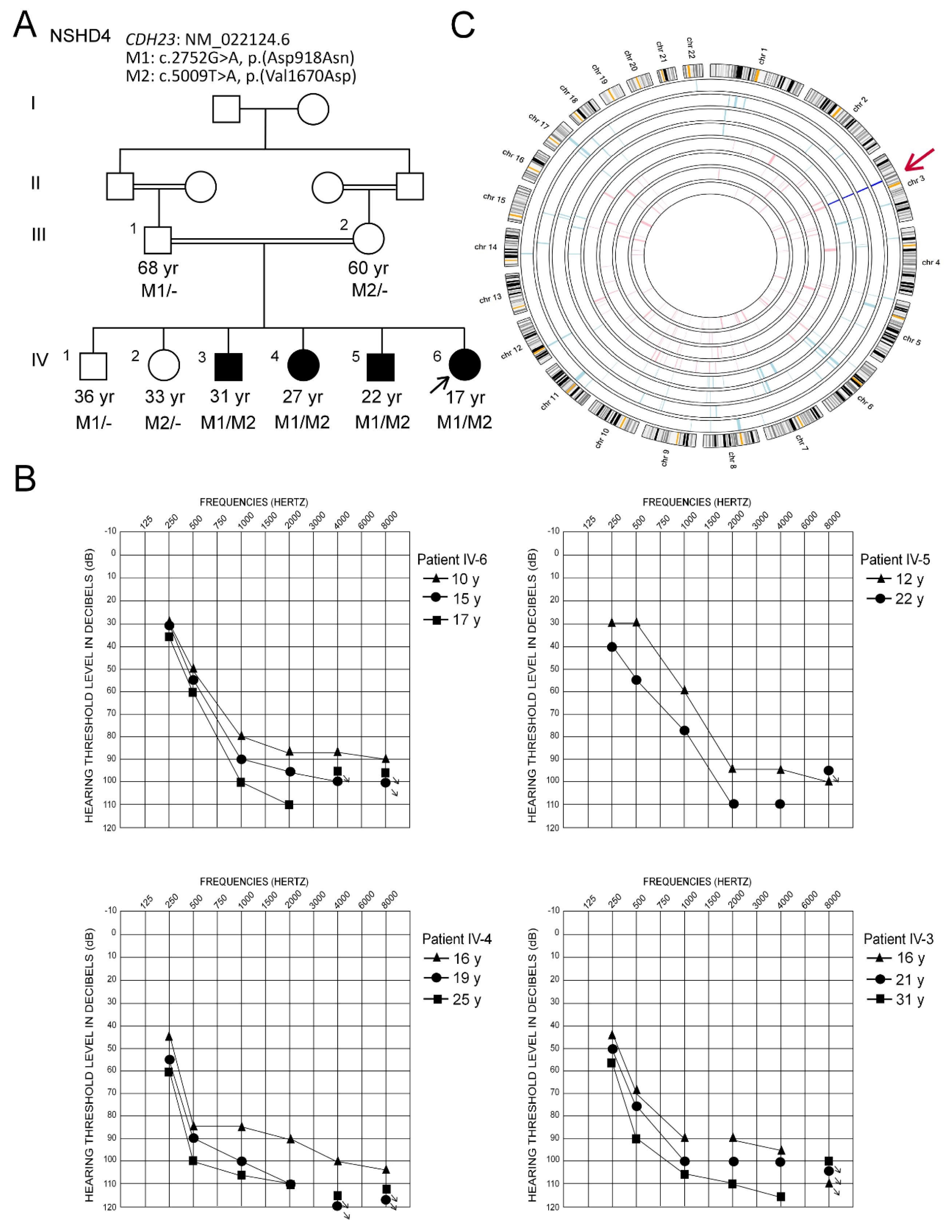
3.1. Clinical Description and Hearing Characteristics
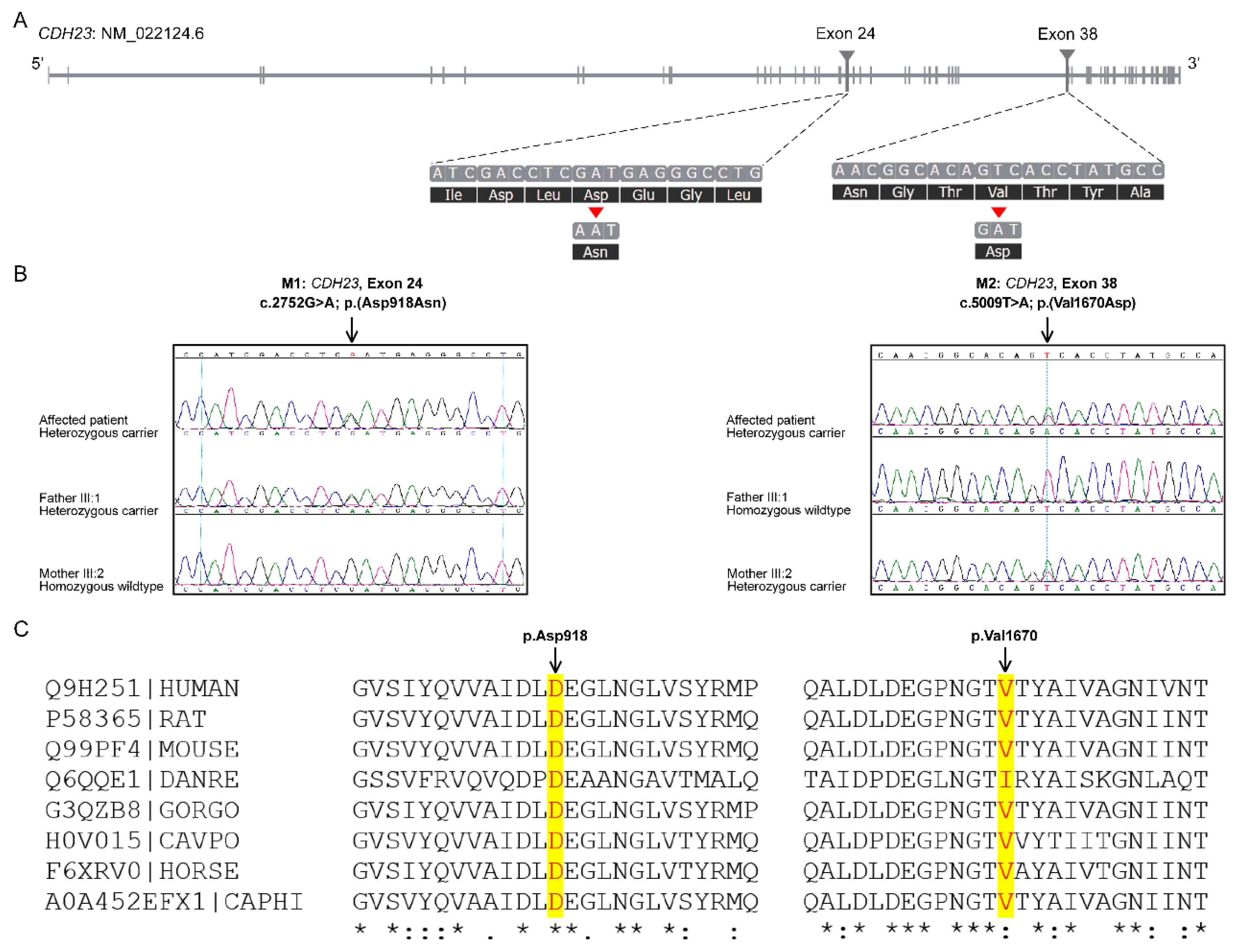
3.2. Genetic Analysis
3.2.1. Autozygome Analysis
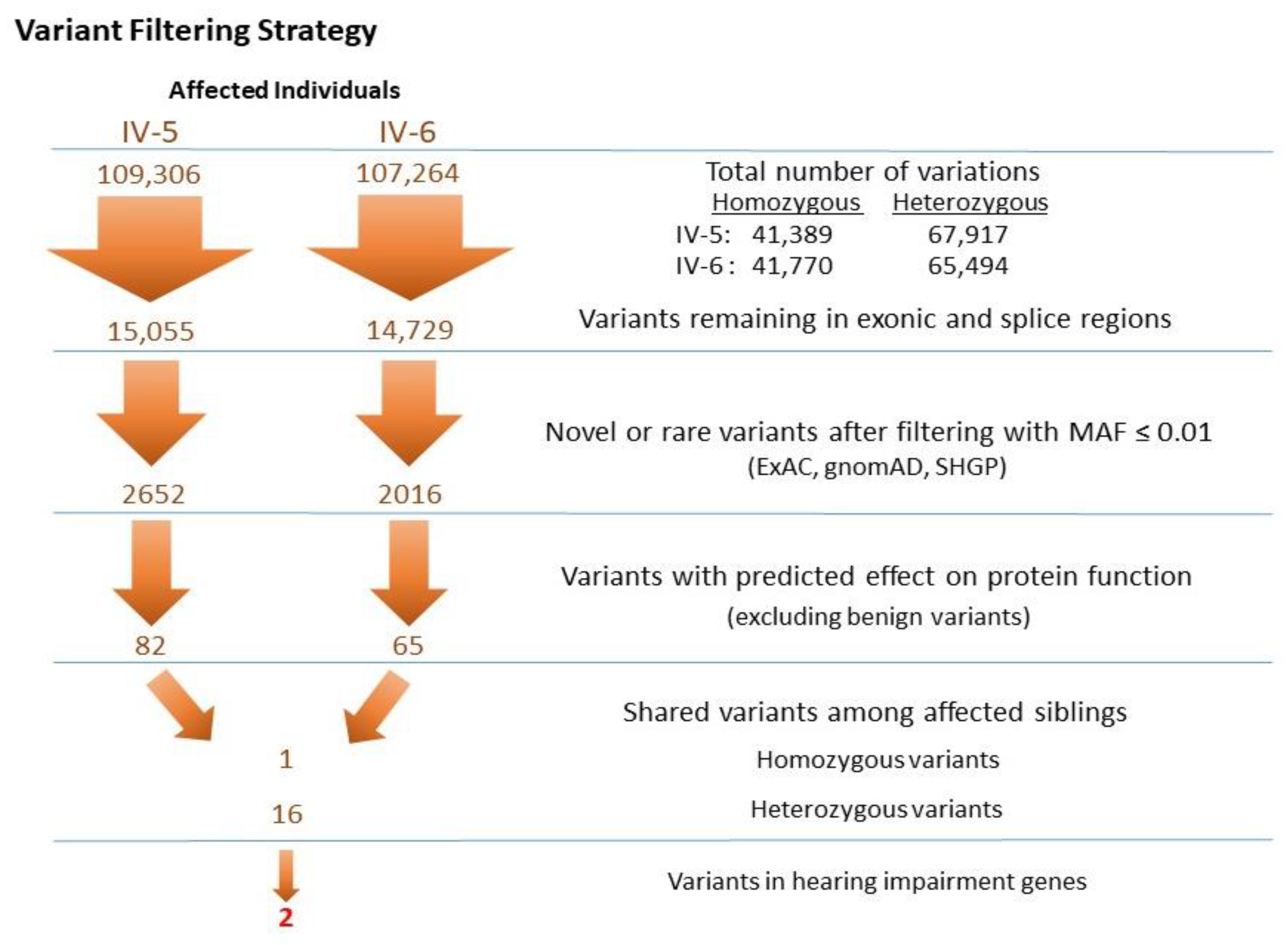
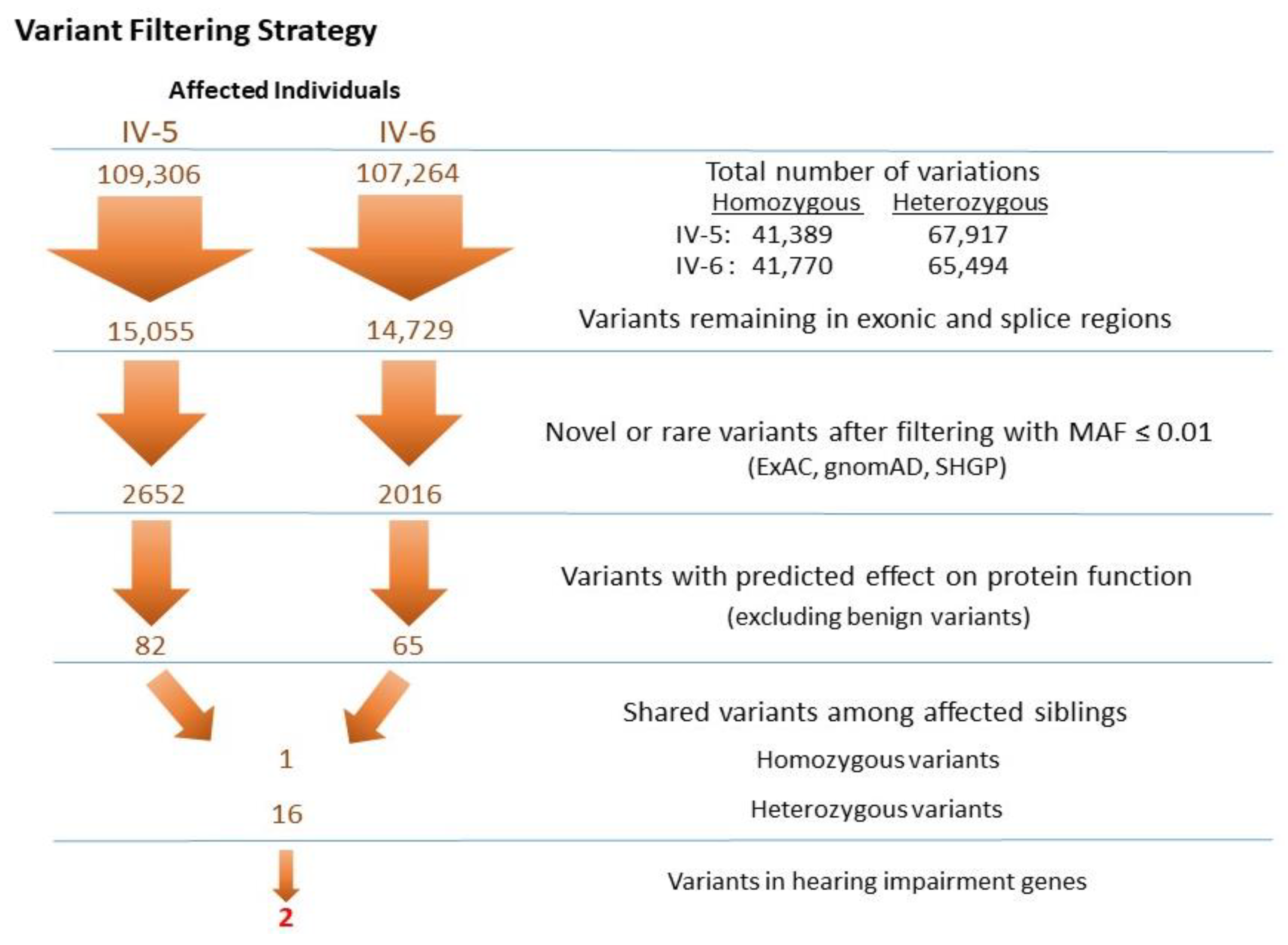
3.2.2. Identification of Mutations by Whole Exome Sequencing
3.2.3. Prediction of the Pathogenic Significance of the Mutations
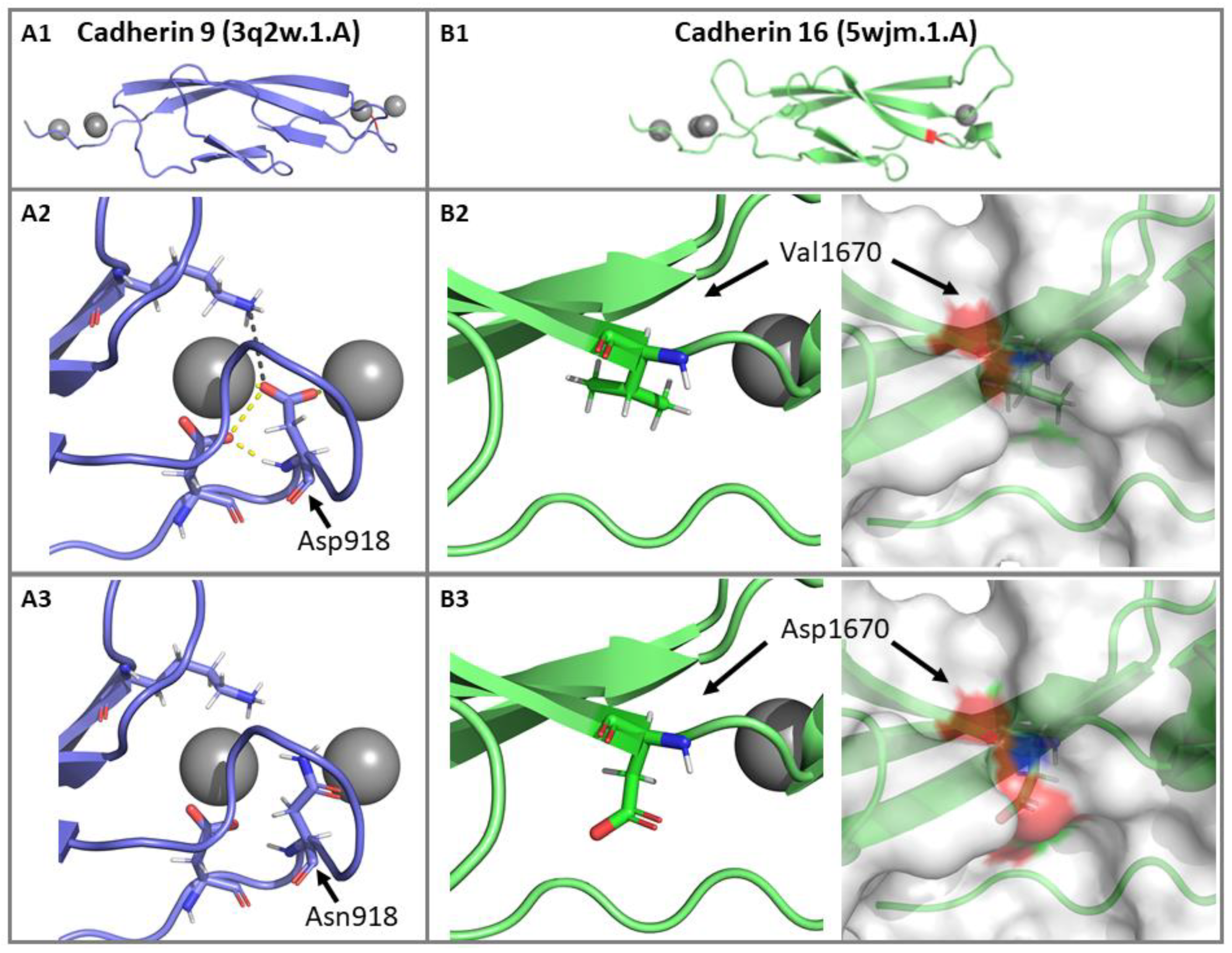
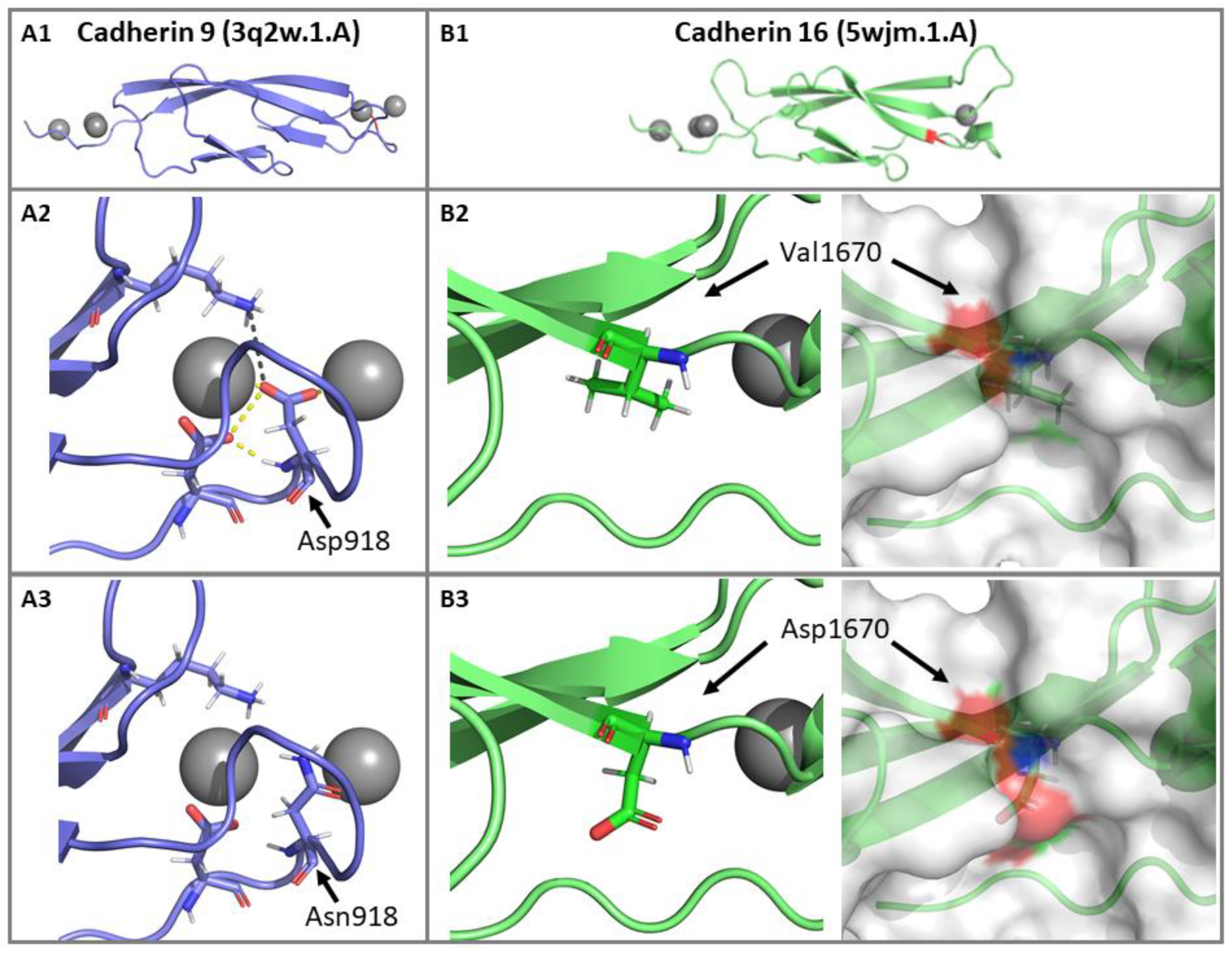
3.2.4. Impact of p.(Asp918Asn) and p.(Val1670Asp) Mutations on the CDH23 3D Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Web Resources
References
- Marazita, M.L.; Ploughman, L.M.; Rawlings, B.; Remington, E.; Arnos, K.S.; Nance, W.E. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am. J. Med Genet. 1993, 46, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.E. Genetic Epidemiology of Hearing Impairment. Ann. N. Y. Acad. Sci. 1991, 630, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Bale, J.F., Jr.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Alford, R.L.; Arnos, K.S.; Fox, M.; Lin, J.W.; Palmer, C.G.; Pandya, A.; Rehm, H.L.; Robin, N.H.; Scott, D.A.; Yoshinaga-Itano, C. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. Off. J. Am. Coll. Med Genet. 2014, 16, 347–355. [Google Scholar]
- Toriello, H.V.S.S. Hereditary Hearing Loss and Its Syndromes, 3rd ed.; Oxford University Press: New York, NY, USA, 2013. [Google Scholar]
- Astuto, L.M.; Bork, J.M.; Weston, M.D.; Askew, J.W.; Fields, R.R.; Orten, D.J.; Ohliger, S.J.; Riazuddin, S.; Morell, R.J.; Khan, S.; et al. CDH23 mutation and phenotype heterogeneity: A profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am. J. Hum. Genet. 2002, 71, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Bolz, H.; Von Brederlow, B.; Ramírez, A.; Bryda, E.C.; Kutsche, K.; Nothwang, H.G.; Seeliger, M.; Cabrera, M.D.C.-S.; Vila, M.C.; Molina, O.P.; et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 2001, 27, 108–112. [Google Scholar] [CrossRef]
- Bork, J.M.; Peters, L.M.; Riazuddin, S.; Bernstein, S.L.; Ahmed, Z.M.; Ness, S.L.; Polomeno, R.; Ramesh, A.; Schloss, M.; Srisailpathy, C.R.S.; et al. Usher Syndrome 1D and Nonsyndromic Autosomal Recessive Deafness DFNB12 Are Caused by Allelic Mutations of the Novel Cadherin-Like Gene CDH. Am. J. Hum. Genet. 2001, 68, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Bork, J.M.; Morell, R.J.; Khan, S.; Riazuddin, S.; Wilcox, E.R.; Friedman, T.B.; Griffith, A.J. Clinical presentation of DFNB12 and Usher syndrome type 1D. Adv. Oto-Rhino-Laryngol. 2002, 61, 145–152. [Google Scholar]
- Friedman, T.B.; Schultz, J.M.; Ahmed, Z.M.; Tsilou, E.T.; Brewer, C. Usher Syndrome: Hearing Loss with Vision Loss. Gene Ther. Cochlear Deaf. 2011, 70, 56–65. [Google Scholar] [CrossRef]
- Oshima, A.; Jaijo, T.; Aller, E.; Millan, J.; Carney, C.; Usami, S.; Moller, C.; Kimberling, W. Mutation profile of theCDH23 gene in 56 probands with Usher syndrome type I. Hum. Mutat. 2008, 29, E37–E46. [Google Scholar] [CrossRef] [Green Version]
- Bolz, H.J.; Roux, A.F. Clinical utility gene card for: Usher syndrome. Eur. J. Hum. Genet. EJHG 2011, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Frolenkov, G.I.; Belyantseva, I.A.; Friedman, T.B.; Griffith, A.J. Genetic insights into the morphogenesis of inner ear hair cells. Nat. Rev. Genet. 2004, 5, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Schmidt, T.; Bienert, S.; Mariani, V.; Studer, G.; Haas, J.; Johner, N.; Schenk, A.D.; Philippsen, A.; Schwede, T. OpenStructure: An integrated software framework for computational structural biology. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Ganapathy, A.; Pandey, N.; Srisailapathy, C.R.S.; Jalvi, R.; Malhotra, V.; Venkatappa, M.; Chatterjee, A.; Sharma, M.; Santhanam, R.; Chadha, S.; et al. Non-Syndromic Hearing Impairment in India: High Allelic Heterogeneity among Mutations in TMPRSS3, TMC1, USHIC, CDH23 and TMIE. PLoS ONE 2014, 9, e84773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botstein, D.; Risch, N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003, 33, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Alkuraya, F.S. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protoc. Hum. Genet. 2012, 6, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, K.; Taibah, K.; Tahir, A.I.; Al Tassan, N.; Berhan, A.; Khater, A.M.; Al-Hazzaa, S.A.; Al-Owain, M.; Imtiaz, F. ILDR1: Novel mutation and a rare cause of congenital deafness in the Saudi Arabian population. Eur. J. Med. Genet. 2014, 57, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, K.; Al-Owain, M.; Al-Numair, N.S.; Afzal, S.; Al-Ageel, S.; Al-Amer, S.; Al-Baik, L.; Al-Otaibi, G.F.; Hashem, A.; Al-Mashharawi, E.; et al. Identification of TMC1 as a relatively common cause for nonsyndromic hearing loss in the Saudi population. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2019, 183, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Miano, M.G.; Jacobson, S.G.; Carothers, A.; Hanson, I.; Teague, P.; Lovell, J.; Cideciyan, A.V.; Haider, N.; Stone, E.M.; Sheffield, V.C.; et al. Pitfalls in homozygosity mapping. Am. J. Hum. Genet. 2000, 67, 1348–1351. [Google Scholar] [CrossRef]
- Ramzan, K.; Al-Owain, M.; Huma, R.; Al-Hazzaa, S.A.; Al-Ageel, S.; Imtiaz, F.; Al-Sayed, M. Utility of whole exome sequencing in the diagnosis of Usher syndrome: Report of novel compound heterozygous MYO7A mutations. Int. J. Pediatr. Otorhinolaryngol. 2018, 108, 17–21. [Google Scholar] [CrossRef]
- Angst, B.D.; Marcozzi, C.; I Magee, A. The cadherin superfamily: Diversity in form and function. J. Cell Sci. 2001, 114, 629–641. [Google Scholar]
- Rowlands, T.M.; Symonds, J.M.; Farookhi, R.; Blaschuk, O.W. Cadherins: Crucial regulators of structure and function in reproductive tissues. Rev. Reprod. 2000, 5, 53–61. [Google Scholar] [CrossRef]
- Nollet, F.; Kools, P.F.J.; Van Roy, F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members 1 1Edited by M. Yaniv. J. Mol. Biol. 2000, 299, 551–572. [Google Scholar] [CrossRef]
- Yang, T.; Wei, X.; Chai, Y.; Li, L.; Wu, H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J. Rare Dis. 2013, 8, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Chaib, H.; Place, C.; Salem, N.; Dodé, C.; Chardenoux, S.; Weissenbach, J.; El Zir, E.; Loiselet, J.; Petit, C. Mapping of DFNB12, a gene for a non-syndromal autosomal recessive deafness, to chromosome 10q21-22. Hum. Mol. Genet. 1996, 5, 1061–1064. [Google Scholar] [CrossRef] [Green Version]
- Wayne, S.; Der Kaloustian, V.M.; Schloss, M.; Polomeno, R.; Scott, D.A.; Hejtmancik, J.F.; Sheffield, V.C.; Smith, R.J.H. Localization of the Usher syndrome type ID gene (Ush1D) to chromosome 10. Hum. Mol. Genet. 1996, 5, 1689–1692. [Google Scholar] [CrossRef] [Green Version]
- Mizutari, K.; Mutai, H.; Namba, K.; Miyanaga, Y.; Nakano, A.; Arimoto, Y.; Masuda, S.; Morimoto, N.; Sakamoto, H.; Kaga, K.; et al. High prevalence of CDH23 mutations in patients with congenital high-frequency sporadic or recessively inherited hearing loss. Orphanet J. Rare Dis. 2015, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennings, R.; Topsakal, V.; Astuto, L.; De Brouwer, A.P.M.; Wagenaar, M.; Huygen, P.L.M.; Kimberling, W.J.; Deutman, A.F.; Kremer, H.; Cremers, C.W.R.J. Variable Clinical Features in Patients with CDH23 Mutations (USH1D-DFNB12). Otol. Neurotol. 2004, 25, 699–706. [Google Scholar] [CrossRef]
- Schultz, J.M.; Bhatti, R.; Madeo, A.C.; Turriff, A.; A Muskett, J.; Zalewski, C.K.; A King, K.; Ahmed, Z.M.; Riazuddin, S.; Ahmad, N.; et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J. Med. Genet. 2011, 48, 767–775. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, A.R.; Lee, C.; Kim, S.Y.; Kim, N.K.D.; Chang, M.Y.; Rhee, J.; Park, M.-H.; Koo, S.K.; Kim, M.Y.; et al. Discovery of CDH23 as a Significant Contributor to Progressive Postlingual Sensorineural Hearing Loss in Koreans. PLoS ONE 2016, 11, e0165680. [Google Scholar] [CrossRef]
- Miyagawa, M.; Nishio, S.Y.; Usami, S. Prevalence and clinical features of hearing loss patients with CDH23 mutations: A large cohort study. PLoS ONE 2012, 7, e40366. [Google Scholar] [CrossRef] [Green Version]
- Müller, U. Cadherins and mechanotransduction by hair cells. Curr. Opin. Cell Biol. 2008, 20, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Siemens, J.; Lillo, C.; Dumont, R.A.; Reynolds, A.; Williams, D.S.; Gillespie, P.G.; Müller, U. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature 2004, 428, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Householder, D.B.; Coppola, V.; Tessarollo, L.; Fritzsch, B.; Lee, E.-C.; Goss, D.; Carlson, G.A.; Copeland, N.G.; Jenkins, N.A. Mutations in Cdh23 Cause Nonsyndromic Hearing Loss in waltzer Mice. Genomics 2001, 74, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, P.; Sakaguchi, H.; Tokita, J.; Wilson-Kubalek, E.M.; Milligan, R.A.; Müller, U.; Kachar, B. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nat. Cell Biol. 2007, 449, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Alagramam, K.N.; Goodyear, R.J.; Geng, R.; Furness, D.N.; Van Aken, A.F.J.; Marcotti, W.; Kros, C.J.; Richardson, G.P. Mutations in Protocadherin 15 and Cadherin 23 Affect Tip Links and Mechanotransduction in Mammalian Sensory Hair Cells. PLoS ONE 2011, 6, e19183. [Google Scholar] [CrossRef]
- Di Palma, F.; Holme, R.H.; Bryda, E.C.; Belyantseva, I.A.; Pellegrino, R.; Kachar, B.; Steel, K.P.; Noben-Trauth, K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 2001, 27, 103–107. [Google Scholar] [CrossRef]
- Holme, R.H.; Steel, K.P. Stereocilia defects in waltzer (Cdh23), shaker1 (Myo7a) and double waltzer/shaker1 mutant mice. Hear. Res. 2002, 169, 13–23. [Google Scholar] [CrossRef]
- Schwander, M.; Xiong, W.; Tokita, J.; Lelli, A.; Elledge, H.M.; Kazmierczak, P.; Sczaniecka, A.; Kolatkar, A.; Wiltshire, T.; Kuhn, P.; et al. A mouse model for nonsyndromic deafness (DFNB12) links hearing loss to defects in tip links of mechanosensory hair cells. Proc. Natl. Acad. Sci. USA 2009, 106, 5252–5257. [Google Scholar] [CrossRef] [Green Version]
- Kane, K.L.; Longo-Guess, C.M.; Gagnon, L.H.; Ding, D.; Salvi, R.J.; Johnson, K.R. Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hear. Res. 2012, 283, 80–88. [Google Scholar] [CrossRef] [Green Version]
- McHugh, R.K.; Friedman, R.A. Genetics of hearing loss: Allelism and modifier genes produce a phenotypic continuum. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2006, 288, 370–381. [Google Scholar] [CrossRef]
- Davis, R.R.; Newlander, J.; Ling, X.-B.; Cortopassi, G.A.; Krieg, E.F.; Erway, L.C. Genetic basis for susceptibility to noise-induced hearing loss in mice. Hear. Res. 2001, 155, 82–90. [Google Scholar] [CrossRef]
- Noben-Trauth, K.; Zheng, Q.Y.; Johnson, K.R. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat. Genet. 2003, 35, 21–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, T.J.; Pawelczyk, M.; Rajkowska, E.; Dudarewicz, A.; Sliwinska-Kowalska, M. Genetic variants of CDH23 associated with noise-induced hearing loss. Otol. Neurotol. 2014, 35, 358–365. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Coordinates and nomenclature | CDH23 (10q22.1) | ||
| Variant 1 | Variant 2 | ||
| Genomic Position (hg19/GRCh37) | chr10:73464686 | chr10:73537600 | |
| dbSNP ID | rs769870573 | rs397517333 | |
| HGVS RefSeq:NM_022124.6 | exon 24 of 70 c.2752G>A, p.(Asp918Asn) | exon 38 of 70 c.5009T>A, p.(Val1670Asp) | |
| Global minor allele frequency (MAF) | GnomAD_exomes | A = 0.000008 (2/246770) | A = 0.000068 (17/249302) |
| ExAC | A = 0.000017 (2/117490) | A = 0.000058 (7/120554) | |
| SHGP | 0 | 0 | |
| In silico pathogenicity prediction tool | CADD Score a | Pathogenic (28.3) | Pathogenic (24.5) |
| PolyPhen-2 | Probably damaging (0.999) | Possibly damaging (0.735) | |
| SIFT | Deleterious (0) | Deleterious (0) | |
| MutationTaster | Disease causing (1) | Disease causing (0.9954) | |
| Mutation assessor | High (3.985) | High (3.82) | |
| DANN | Pathogenic (0.9991) | Pathogenic (0.9807) | |
| FATHMM-MKL | Damaging (0.9939) | Damaging (0.9695) | |
| MetaSVM | Damaging (0.3538) | Damaging (0.5357) | |
| MetaLR | Damaging (0.5892) | Damaging (0.6209) | |
| Conservation GERP++ b | Conserved (5.4) | Conserved (5.76) | |
| ACMG variant classificationc | PM1, PM2, PP1, PP2, PP3 | PM2, PP1, PP2, PP3 | |
| “Pathogenic” | “Likely Pathogenic” | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramzan, K.; Al-Numair, N.S.; Al-Ageel, S.; Elbaik, L.; Sakati, N.; Al-Hazzaa, S.A.F.; Al-Owain, M.; Imtiaz, F. Identification of Novel CDH23 Variants Causing Moderate to Profound Progressive Nonsyndromic Hearing Loss. Genes 2020, 11, 1474. https://doi.org/10.3390/genes11121474
Ramzan K, Al-Numair NS, Al-Ageel S, Elbaik L, Sakati N, Al-Hazzaa SAF, Al-Owain M, Imtiaz F. Identification of Novel CDH23 Variants Causing Moderate to Profound Progressive Nonsyndromic Hearing Loss. Genes. 2020; 11(12):1474. https://doi.org/10.3390/genes11121474
Chicago/Turabian StyleRamzan, Khushnooda, Nouf S. Al-Numair, Sarah Al-Ageel, Lina Elbaik, Nadia Sakati, Selwa A. F. Al-Hazzaa, Mohammed Al-Owain, and Faiqa Imtiaz. 2020. "Identification of Novel CDH23 Variants Causing Moderate to Profound Progressive Nonsyndromic Hearing Loss" Genes 11, no. 12: 1474. https://doi.org/10.3390/genes11121474
APA StyleRamzan, K., Al-Numair, N. S., Al-Ageel, S., Elbaik, L., Sakati, N., Al-Hazzaa, S. A. F., Al-Owain, M., & Imtiaz, F. (2020). Identification of Novel CDH23 Variants Causing Moderate to Profound Progressive Nonsyndromic Hearing Loss. Genes, 11(12), 1474. https://doi.org/10.3390/genes11121474