The Genetic Basis of Obesity and Related Metabolic Diseases in Humans and Companion Animals



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Factors Contributing to Obesity
Obesity Susceptibility Is Highly Heritable
3. Studies of Monogenic Obesity Have Been Highly Informative
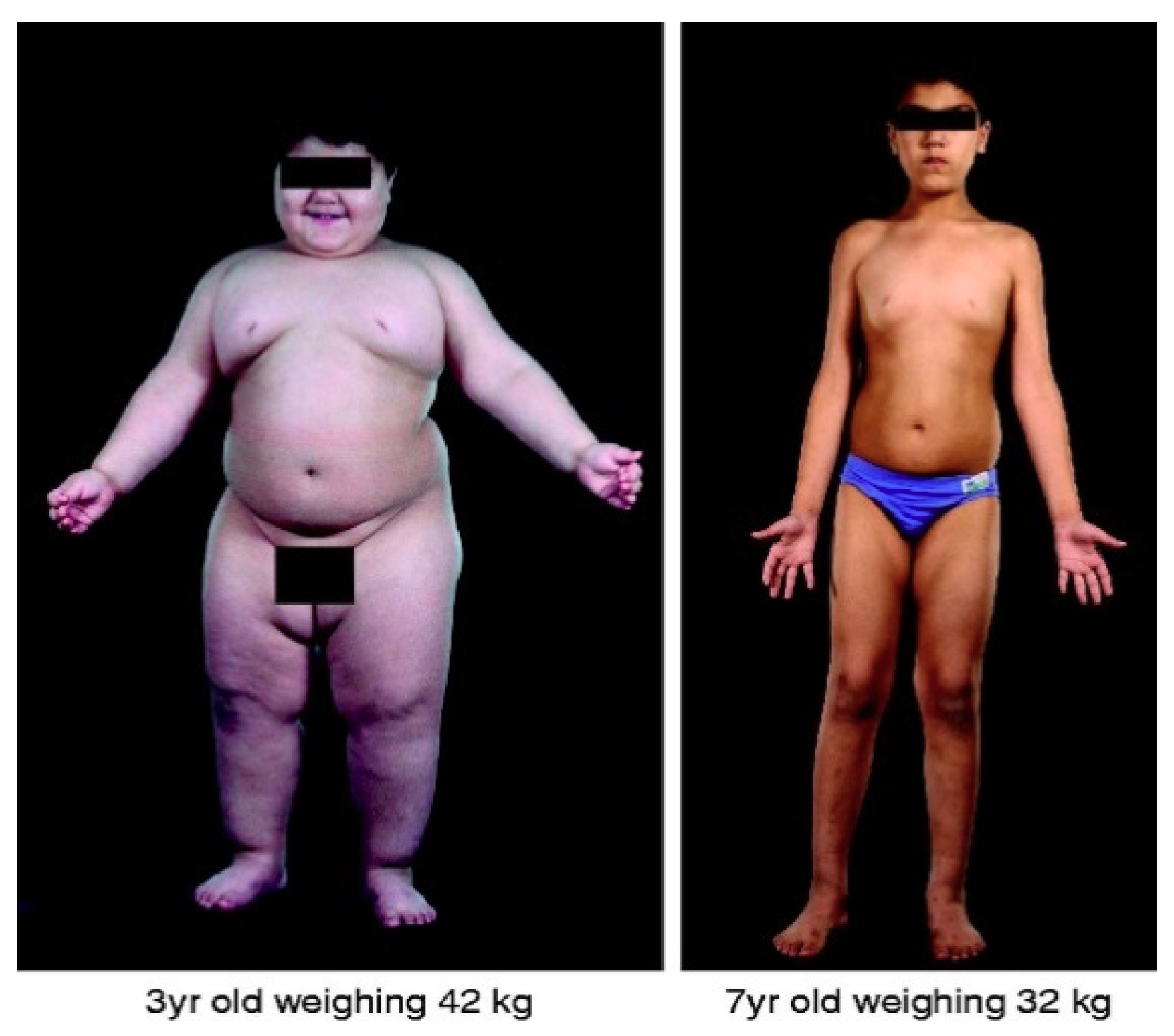
3.1. The Discovery of Leptin
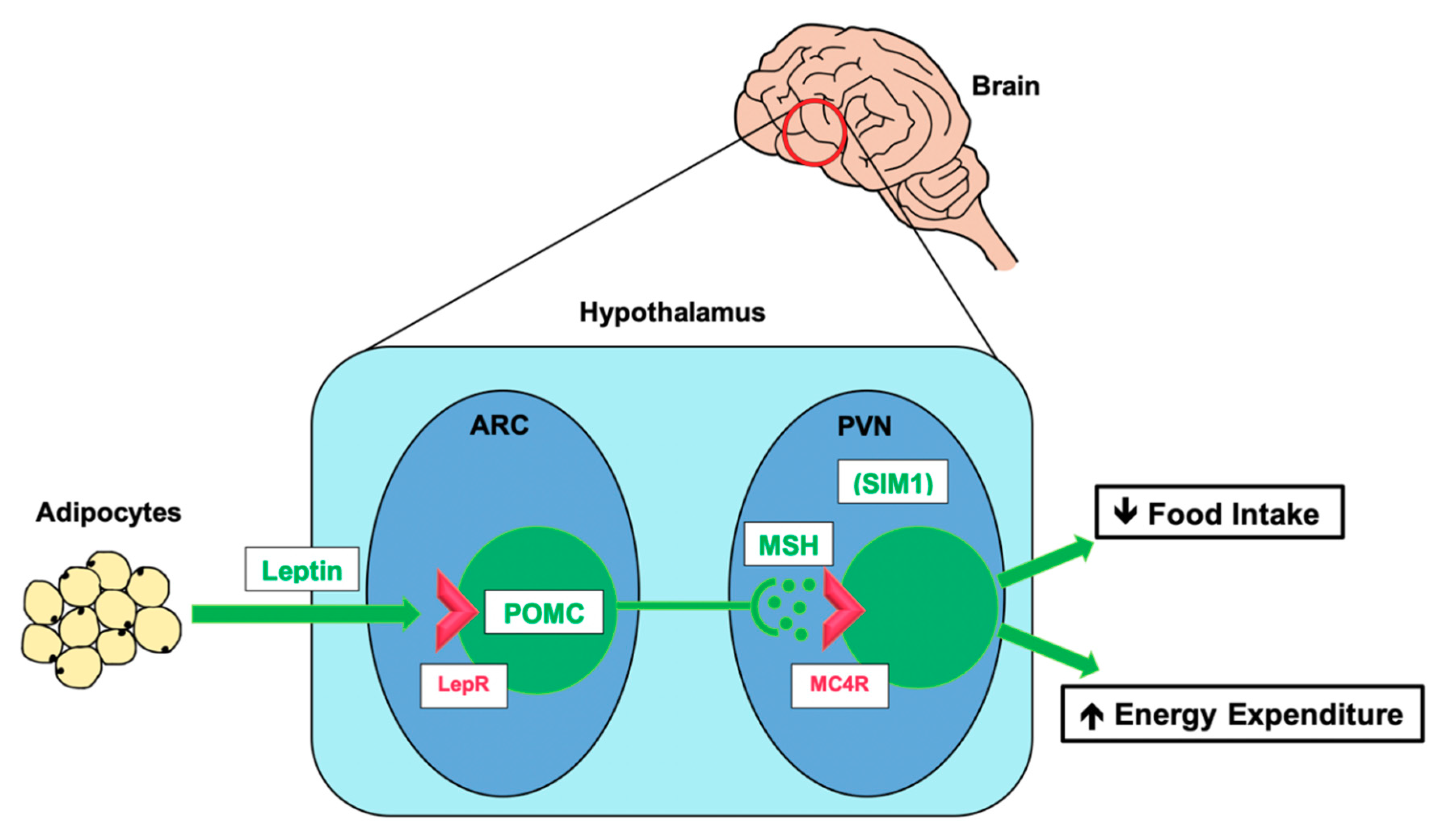
3.2. The Leptin–Melanocortin Pathway
3.3. Disruption of Leptin–Melanocortin Signalling Leads to Obesity
3.4. Other Causes of Monogenic Obesity
4. Common Human Obesity
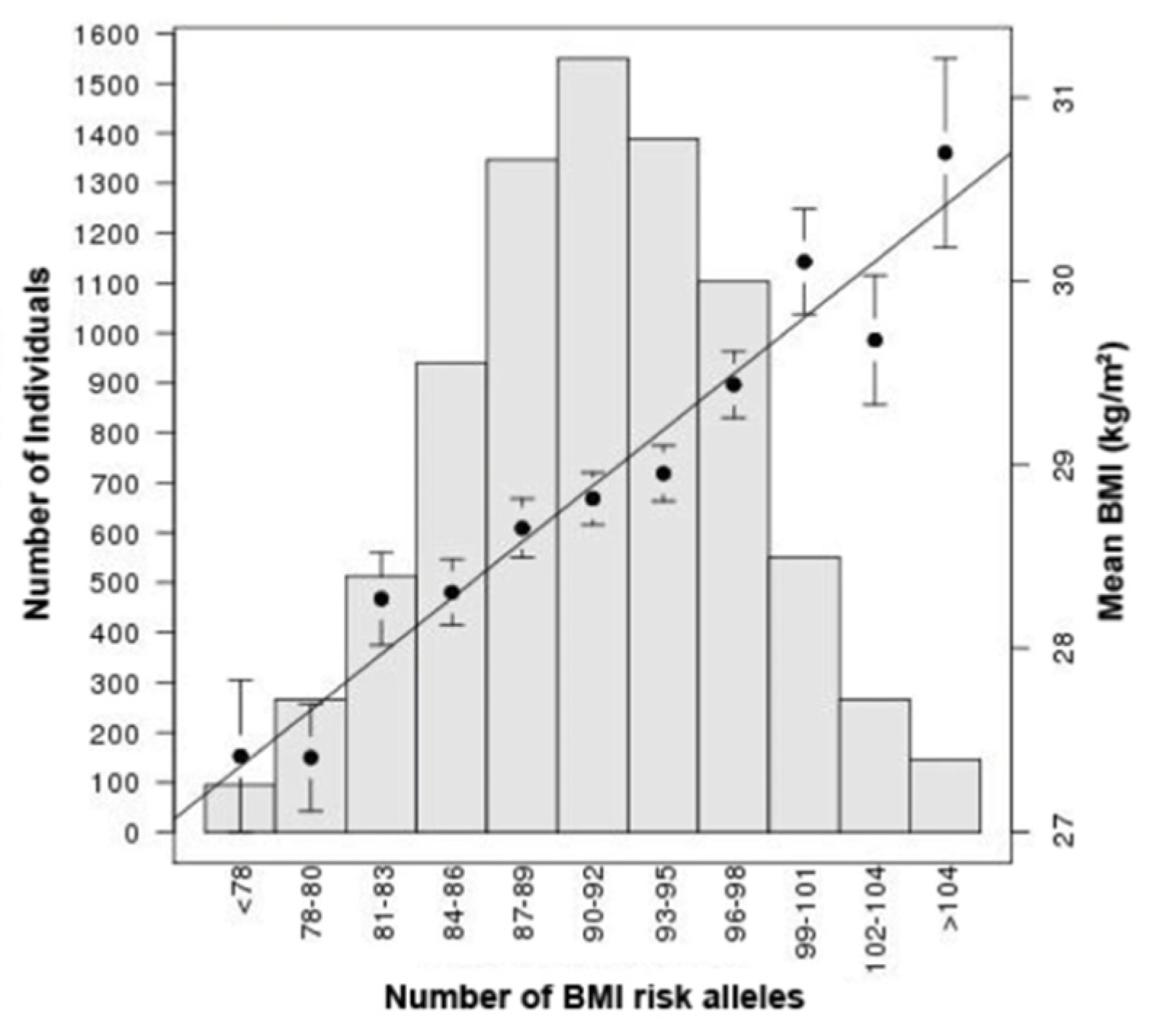
From GWAS to Function
5. Genetic Insight into Obesity Comorbidities and Metabolic Syndrome
Overview—Molecular Mechanisms Underlying Obesity Co-Morbidities
6. Applying Current Knowledge to Study Companion Animal Disease
7. Canine Obesity Genetics
7.1. Genes Investigated in Canine Obesity
7.1.1. POMC
7.1.2. MC4R
7.1.3. FTO
7.1.4. MC3R
7.1.5. INSIG2
7.1.6. GPR120/FFAR4
7.1.7. PPARs
7.1.8. Adipokines
8. Feline Obesity and Associated Disease
8.1. Evidence for the Role of Genetics in Feline Obesity and Related Disease
8.2. Familial Obesity in a Feline Colony
8.3. Genetics of Diabetes Mellitus in Pet Cats
9. Obesity and Related Metabolic Disease in Horses
9.1. Genetics Influence Equine Obesity, EMS, and Laminitis
9.2. GWAS for EMS and Related Traits
10. From Humans to Animals and Back Again
10.1. Lessons for Animal Genetics
10.2. Lessons from Animal Genetics
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight. Global Health Observatory (GHO) Data. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 21 June 2020).
- Cave, N.J.; Allan, F.J.; Schokkenbroek, S.L.; Metekohy, C.A.; Pfeiffer, D.U. A cross-sectional study to compare changes in the prevalence and risk factors for feline obesity between 1993 and 2007 in New Zealand. Prev. Vet. Med. 2012, 107, 121–133. [Google Scholar] [CrossRef]
- Courcier, E.A.; Thomson, R.M.; Mellor, D.J.; Yam, P.S. An epidemiological study of environmental factors associated with canine obesity. J. Small Anim. Pract. 2010, 51, 362–367. [Google Scholar] [CrossRef]
- World Health Organisation. Obesity: Preventing and managing the global epidemic. WHO Consult. 2000, 894, 1–253. [Google Scholar]
- BSAVA. Obesity. 2019. Available online: https://bit.ly/2Pb9oRa (accessed on 4 September 2020).
- Farooqi, S.; O’Rahilly, S. Genetics of obesity in humans. Endocr. Rev. 2006, 27, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Raffan, E.; Dennis, R.J.; O’Donovan, C.J.; Becker, J.M.; Scott, R.A.; Smith, S.P.; Withers, D.J.; Wood, C.J.; Conci, E.; Clements, D.N.; et al. A Deletion in the Canine POMC Gene Is Associated with Weight and Appetite in Obesity-Prone Labrador Retriever Dogs. Cell Metab. 2016, 23, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Paeratakul, S.; Lovejoy, J.C.; Ryan, D.H.; Bray, G.A. The relation of gender, race and socioeconomic status to obesity and obesity comorbidities in a sample of US adults. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1205–1210. [Google Scholar] [CrossRef]
- McGreevy, P.D.; Thomson, P.C.; Pride, C.; Fawcett, A.; Grassi, T.; Jones, B. Prevalence of obesity in dogs examined by Australian veterinary practices and the risk factors involved. Vet. Rec. 2005, 156, 695–702. [Google Scholar] [CrossRef]
- Lund, E.M.; Armstrong, P.J.; Kirk, C.A.; Klausner, J.S. Prevalence and Risk Factors for Obesity in Adult Dogs from Private US Veterinary Practices. Int. J. Appl. Res. Vet. Med. 2006, 4, 177–186. [Google Scholar]
- Robin, C.A.; Ireland, J.L.; Wylie, C.E.; Collins, S.N.; Verheyen, K.L.; Newton, J.R. Prevalence of and risk factors for equine obesity in Great Britain based on owner-reported body condition scores. Equine Vet. J. 2015, 47, 196–201. [Google Scholar] [CrossRef]
- Lund, E.M.; Armstrong, P.J.; Kirk, C.A.; Klausner, J.S. Prevalence and Risk Factors for Obesity in Adult Cats from Private US Veterinary Practices. Int. J. Appl. Res. Vet. Med. 2005, 3, 88–96. [Google Scholar]
- Brewis, A.; SturtzSreetharan, C.; Wutich, A. Obesity stigma as a globalizing health challenge. Glob. Health 2018, 14, 20. [Google Scholar] [CrossRef]
- Friedman, M. Mother blame, fat shame, and moral panic:“Obesity” and child welfare. Fat Stud. 2015, 4, 14–27. [Google Scholar] [CrossRef]
- Pearl, R.L.; Wadden, T.A.; Bach, C.; Leonard, S.M.; Michel, K.E. Who’s a good boy? Effects of dog and owner body weight on veterinarian perceptions and treatment recommendations. Int. J. Obes. (Lond.) 2020. [Google Scholar] [CrossRef] [PubMed]
- German, A.; Ramsey, I.; Lhermette, P. We should classify pet obesity as a disease. Vet. Rec. 2019, 185, 735. [Google Scholar] [CrossRef] [PubMed]
- van der Klaauw, A.A.; Farooqi, I.S. The hunger genes: Pathways to obesity. Cell 2015, 161, 119–132. [Google Scholar] [CrossRef]
- Silventoinen, K.; Magnusson, P.K.; Tynelius, P.; Kaprio, J.; Rasmussen, F. Heritability of body size and muscle strength in young adulthood: A study of one million Swedish men. Genet. Epidemiol. 2008, 32, 341–349. [Google Scholar] [CrossRef]
- Wardle, J.; Carnell, S.; Haworth, C.M.; Plomin, R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am. J. Clin. Nutr. 2008, 87, 398–404. [Google Scholar] [CrossRef]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef]
- Yen, T.T.; Stienmetz, J.; Simpson, P.J. Blood volume of obese (ob-ob) and diabetic (db-db) mice. Proc. Soc. Exp. Biol. Med. 1970, 133, 307–308. [Google Scholar] [CrossRef]
- Coleman, D.L. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 1973, 9, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Wabitsch, M.; Funcke, J.B.; Lennerz, B.; Kuhnle-Krahl, U.; Lahr, G.; Debatin, K.M.; Vatter, P.; Gierschik, P.; Moepps, B.; Fischer-Posovszky, P. Biologically inactive leptin and early-onset extreme obesity. N. Engl. J. Med. 2015, 372, 48–54. [Google Scholar] [CrossRef]
- Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998, 18, 213–215. [Google Scholar] [CrossRef]
- Zhao, Y.; Hong, N.; Liu, X.; Wu, B.; Tang, S.; Yang, J.; Hu, C.; Jia, W. A novel mutation in leptin gene is associated with severe obesity in Chinese individuals. Biomed. Res. Int. 2014, 2014, 912052. [Google Scholar] [CrossRef]
- Clement, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Wangensteen, T.; Collins, S.; Kimber, W.; Matarese, G.; Keogh, J.M.; Lank, E.; Bottomley, B.; Lopez-Fernandez, J.; Ferraz-Amaro, I.; et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med. 2007, 356, 237–247. [Google Scholar] [CrossRef]
- Hannema, S.E.; Wit, J.M.; Houdijk, M.E.; van Haeringen, A.; Bik, E.C.; Verkerk, A.J.; Uitterlinden, A.G.; Kant, S.G.; Oostdijk, W.; Bakker, E.; et al. Novel Leptin Receptor Mutations Identified in Two Girls with Severe Obesity Are Associated with Increased Bone Mineral Density. Horm. Res. Paediatr. 2016, 85, 412–420. [Google Scholar] [CrossRef]
- Farr, O.M.; Gavrieli, A.; Mantzoros, C.S. Leptin applications in 2015: What have we learned about leptin and obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Dubern, B.; Clement, K. Leptin and leptin receptor-related monogenic obesity. Biochimie 2012, 94, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Nunziata, A.; Funcke, J.B.; Borck, G.; von Schnurbein, J.; Brandt, S.; Lennerz, B.; Moepps, B.; Gierschik, P.; Fischer-Posovszky, P.; Wabitsch, M. Functional and Phenotypic Characteristics of Human Leptin Receptor Mutations. J. Endocr. Soc. 2019, 3, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; O’Rahilly, S. 20 years of leptin: Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef]
- Garfield, A.S.; Lam, D.D.; Marston, O.J.; Przydzial, M.J.; Heisler, L.K. Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol. Metab. 2009, 20, 203–215. [Google Scholar] [CrossRef]
- Barr, V.A.; Malide, D.; Zarnowski, M.J.; Taylor, S.I.; Cushman, S.W. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology 1997, 138, 4463–4472. [Google Scholar] [CrossRef]
- Amitani, M.; Asakawa, A.; Amitani, H.; Inui, A. The role of leptin in the control of insulin-glucose axis. Front. Neurosci. 2013, 7, 51. [Google Scholar] [CrossRef]
- Cummings, D.E.; Schwartz, M.W. Genetics and pathophysiology of human obesity. Annu. Rev. Med. 2003, 54, 453–471. [Google Scholar] [CrossRef]
- Friedman, J. Leading the charge in leptin research: An interview with Jeffrey Friedman. Dis. Model. Mech. 2012, 5, 576–579. [Google Scholar]
- Oswal, A.; Yeo, G.S. The leptin melanocortin pathway and the control of body weight: Lessons from human and murine genetics. Obes. Rev. 2007, 8, 293–306. [Google Scholar] [CrossRef]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Krude, H.; Biebermann, H.; Schnabel, D.; Tansek, M.Z.; Theunissen, P.; Mullis, P.E.; Gruters, A. Obesity due to proopiomelanocortin deficiency: Three new cases and treatment trials with thyroid hormone and ACTH4-10. J. Clin. Endocrinol. Metab. 2003, 88, 4633–4640. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Drop, S.; Clements, A.; Keogh, J.M.; Biernacka, J.; Lowenbein, S.; Challis, B.G.; O’Rahilly, S. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes 2006, 55, 2549–2553. [Google Scholar] [CrossRef] [PubMed]
- O’Rahilly, S.; Farooqi, I.S.; Yeo, G.S.; Challis, B.G. Minireview: Human obesity-lessons from monogenic disorders. Endocrinology 2003, 144, 3757–3764. [Google Scholar] [CrossRef] [PubMed]
- Burnett, L.C.; LeDuc, C.A.; Sulsona, C.R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J.F.; Morabito, M.V.; Skowronski, A.A.; Hubner, G.; et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Investig. 2017, 127, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Ramachandrappa, S.; Raimondo, A.; Cali, A.M.; Keogh, J.M.; Henning, E.; Saeed, S.; Thompson, A.; Garg, S.; Bochukova, E.G.; Brage, S.; et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J. Clin. Investig. 2013, 123, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Raimondo, A.; Stutzmann, F.; Ghoussaini, M.; Ramachandrappa, S.; Bersten, D.C.; Durand, E.; Vatin, V.; Balkau, B.; Lantieri, O.; et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. J. Clin. Investig. 2013, 123, 3037–3041. [Google Scholar] [CrossRef]
- Yeo, G.S.; Farooqi, I.S.; Aminian, S.; Halsall, D.J.; Stanhope, R.G.; O’Rahilly, S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat. Genet. 1998, 20, 111–112. [Google Scholar] [CrossRef]
- Vaisse, C.; Clement, K.; Guy-Grand, B.; Froguel, P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 1998, 20, 113–114. [Google Scholar] [CrossRef]
- Kobayashi, H.; Ogawa, Y.; Shintani, M.; Ebihara, K.; Shimodahira, M.; Iwakura, T.; Hino, M.; Ishihara, T.; Ikekubo, K.; Kurahachi, H.; et al. A Novel homozygous missense mutation of melanocortin-4 receptor (MC4R) in a Japanese woman with severe obesity. Diabetes 2002, 51, 243–246. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Yeo, G.S.; Keogh, J.M.; Aminian, S.; Jebb, S.A.; Butler, G.; Cheetham, T.; O’Rahilly, S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Investig. 2000, 106, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Loid, P.; Mustila, T.; Makitie, R.E.; Viljakainen, H.; Kampe, A.; Tossavainen, P.; Lipsanen-Nyman, M.; Pekkinen, M.; Makitie, O. Rare Variants in Genes Linked to Appetite Control and Hypothalamic Development in Early-Onset Severe Obesity. Front. Endocrinol. (Lausanne) 2020, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Doulla, M.; McIntyre, A.D.; Hegele, R.A.; Gallego, P.H. A novel MC4R mutation associated with childhood-onset obesity: A case report. Paediatr. Child Health 2014, 19, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S. Monogenic human obesity. Front. Horm. Res. 2008, 36, 1–11. [Google Scholar]
- Chami, N.; Preuss, M.; Walker, R.W.; Moscati, A.; Loos, R.J.F. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK Biobank population. PLoS Med. 2020, 17, e1003196. [Google Scholar] [CrossRef]
- Loos, R.J.; Lindgren, C.M.; Li, S.; Wheeler, E.; Zhao, J.H.; Prokopenko, I.; Inouye, M.; Freathy, R.M.; Attwood, A.P.; Beckmann, J.S.; et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 2008, 40, 768–775. [Google Scholar] [CrossRef]
- Lotta, L.A.; Mokrosinski, J.; Mendes de Oliveira, E.; Li, C.; Sharp, S.J.; Luan, J.; Brouwers, B.; Ayinampudi, V.; Bowker, N.; Kerrison, N.; et al. Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 2019, 177, 597–607.e9. [Google Scholar] [CrossRef]
- van der Klaauw, A.A.; Croizier, S.; Mendes de Oliveira, E.; Stadler, L.K.J.; Park, S.; Kong, Y.; Banton, M.C.; Tandon, P.; Hendricks, A.E.; Keogh, J.M.; et al. Human Semaphorin 3 Variants Link Melanocortin Circuit Development and Energy Balance. Cell 2019, 176, 729–742.e18. [Google Scholar] [CrossRef]
- Cordeira, J.; Rios, M. Weighing in the role of BDNF in the central control of eating behavior. Mol. Neurobiol. 2011, 44, 441–448. [Google Scholar] [CrossRef]
- Pearce, L.R.; Atanassova, N.; Banton, M.C.; Bottomley, B.; van der Klaauw, A.A.; Revelli, J.P.; Hendricks, A.; Keogh, J.M.; Henning, E.; Doree, D.; et al. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell 2013, 155, 765–777. [Google Scholar] [CrossRef]
- Minster, R.L.; Hawley, N.L.; Su, C.T.; Sun, G.; Kershaw, E.E.; Cheng, H.; Buhule, O.D.; Lin, J.; Reupena, M.S.; Viali, S.; et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat. Genet. 2016, 48, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, A.C.P.; Mastronardi, C.; Johar, A.; Arcos-Burgos, M.; Paz-Filho, G. Genetics of non-syndromic childhood obesity and the use of high-throughput DNA sequencing technologies. J. Diabetes Complicat. 2017, 31, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.; Lee, S.H.; Robinson, M.R.; Perry, J.R.; Nolte, I.M.; van Vliet-Ostaptchouk, J.V.; et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 2015, 47, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Go, M.J.; Kim, Y.J.; Heo, J.Y.; Oh, J.H.; Ban, H.J.; Yoon, D.; Lee, M.H.; Kim, D.J.; Park, M.; et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat. Genet. 2009, 41, 527–534. [Google Scholar] [CrossRef]
- Pulit, S.L.; Stoneman, C.; Morris, A.P.; Wood, A.R.; Glastonbury, C.A.; Tyrrell, J.; Yengo, L.; Ferreira, T.; Marouli, E.; Ji, Y.; et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019, 28, 166–174. [Google Scholar] [CrossRef]
- Lotta, L.A.; Gulati, P.; Day, F.R.; Payne, F.; Ongen, H.; van de Bunt, M.; Gaulton, K.J.; Eicher, J.D.; Sharp, S.J.; Luan, J.; et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 2017, 49, 17–26. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Karlsson, T.; Ek, W.E.; Johansson, A. Genome-wide association study of body fat distribution identifies adiposity loci and sex-specific genetic effects. Nat. Commun. 2019, 10, 339. [Google Scholar] [CrossRef]
- Hebebrand, J.; Volckmar, A.L.; Knoll, N.; Hinney, A. Chipping away the ‘missing heritability’: GIANT steps forward in the molecular elucidation of obesity–but still lots to go. Obes. Facts 2010, 3, 294–303. [Google Scholar] [CrossRef]
- Llewellyn, C.H.; Trzaskowski, M.; Plomin, R.; Wardle, J. Finding the missing heritability in pediatric obesity: The contribution of genome-wide complex trait analysis. Int. J. Obes. (Lond.) 2013, 37, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.J. Genetic determinants of common obesity and their value in prediction. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Lindgren, C.M. The genetics of obesity. Curr. Diabetes Rep. 2010, 10, 498–505. [Google Scholar] [CrossRef]
- Xia, Q.; Grant, S.F. The genetics of human obesity. Ann. N. Y. Acad. Sci. 2013, 1281, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.K.; Lindblad-Toh, K. Leader of the pack: Gene mapping in dogs and other model organisms. Nat. Rev. Genet. 2008, 9, 713–725. [Google Scholar] [CrossRef]
- Hall, A.B.; Tolonen, A.C.; Xavier, R.J. Human genetic variation and the gut microbiome in disease. Nat. Rev. Genet. 2017, 18, 690–699. [Google Scholar] [CrossRef]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef]
- Lambert, S.A.; Abraham, G.; Inouye, M. Towards clinical utility of polygenic risk scores. Hum. Mol. Genet. 2019, 28, R133–R142. [Google Scholar] [CrossRef]
- Speakman, J.R.; Loos, R.J.F.; O’Rahilly, S.; Hirschhorn, J.N.; Allison, D.B. GWAS for BMI: A treasure trove of fundamental insights into the genetic basis of obesity. Int. J. Obes. (Lond.) 2018, 42, 1524–1531. [Google Scholar] [CrossRef]
- Farooqi, I.S. Chapter 4: Genetics of Obesity. In Handbook of Obesity Treatment; Guilford Publications: New York, NY, USA, 2018; pp. 64–74. [Google Scholar]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef]
- O’Rahilly, S.; Coll, A.P.; Yeo, G.S. FTO Obesity Variant and Adipocyte Browning in Humans. N. Engl. J. Med. 2016, 374, 191. [Google Scholar] [CrossRef] [PubMed]
- Leow, M.K. FTO Obesity Variant and Adipocyte Browning in Humans. N. Engl. J. Med. 2016, 374, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Apovian, C.M. Obesity: Definition, comorbidities, causes, and burden. Am. J. Manag. Care 2016, 22, s176–s185. [Google Scholar]
- Kyrou, I.; Randeva, H.S.; Tsigos, C.; Kaltsas, G.; Weickert, M.O. Clinical Problems Caused by Obesity. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Eds.; Endotext, MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Pi-Sunyer, X. The medical risks of obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Lean, M.E. A clinical perspective of obesity, metabolic syndrome and cardiovascular disease. JRSM Cardiovasc. Dis. 2016, 5, 2048004016633371. [Google Scholar] [CrossRef]
- Stefan, N.; Haring, H.U.; Hu, F.B.; Schulze, M.B. Metabolically healthy obesity: Epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 2013, 1, 152–162. [Google Scholar] [CrossRef]
- Eckel, R.H.; Kahn, S.E.; Ferrannini, E.; Goldfine, A.B.; Nathan, D.M.; Schwartz, M.W.; Smith, R.J.; Smith, S.R.; Endocrine, S.; American Diabetes, A.; et al. Obesity and type 2 diabetes: What can be unified and what needs to be individualized? Diabetes Care 2011, 34, 1424–1430. [Google Scholar] [CrossRef]
- Semple, R.K. How does insulin resistance arise, and how does it cause disease? Human genetic lessons. Eur. J. Endocrinol. 2016, 174, R209–R223. [Google Scholar] [CrossRef]
- World Health Organisation. Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies. WHO Consult. Lancet 2004, 363, 157–163. [Google Scholar]
- Goodarzi, M.O. Genetics of obesity: What genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018, 6, 223–236. [Google Scholar] [CrossRef]
- Romeo, S.; Sanyal, A.; Valenti, L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab. 2020, 31, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Rung, J.; Cauchi, S.; Albrechtsen, A.; Shen, L.; Rocheleau, G.; Cavalcanti-Proenca, C.; Bacot, F.; Balkau, B.; Belisle, A.; Borch-Johnsen, K.; et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat. Genet. 2009, 41, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.L.; Savage, D.B. The genetics of lipid storage and human lipodystrophies. Trends Mol. Med. 2015, 21, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, T.I.A. From fat cells through an obesity theory. Eur. J. Clin. Nutr. 2018, 72, 1329–1335. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—An allostatic perspective. Biochim. Biophys. Acta 2010, 1801, 338–349. [Google Scholar] [CrossRef]
- German, A.J. The growing problem of obesity in dogs and cats. J. Nutr. 2006, 136, 1940S–1946S. [Google Scholar] [CrossRef]
- Lucena, S.; Lamy, E.; Capela, F.; Lavrador, C.; Tvarijonaviciute, A. Human and Canine Prevalence of Obesity and Feeding Habits–a One Health Approach in Portugal. In Proceedings of the Conference Proceedings: ICAAM—Comunicações—Em Congressos Científicos Internacionais, Évora, Portugal, 15–16 October 2018; Available online: http://hdl.handle.net/10174/24279 (accessed on 9 September 2020).
- Colliard, L.; Ancel, J.; Benet, J.J.; Paragon, B.M.; Blanchard, G. Risk Factors for Obesity in Dogs in France. J. Nutr. 2006, 136, 1951S–1954S. [Google Scholar] [CrossRef]
- Mao, J.; Xia, Z.; Chen, J.; Yu, J. Prevalence and risk factors for canine obesity surveyed in veterinary practices in Beijing, China. Prev. Vet. Med. 2013, 112, 438–442. [Google Scholar] [CrossRef]
- German, A.J.; Hervera, M.; Hunter, L.; Holden, S.L.; Morris, P.J.; Biourge, V.; Trayhurn, P. Improvement in insulin resistance and reduction in plasma inflammatory adipokines after weight loss in obese dogs. Domest. Anim. Endocrinol. 2009, 37, 214–226. [Google Scholar] [CrossRef]
- Chandler, M.; Cunningham, S.; Lund, E.M.; Khanna, C.; Naramore, R.; Patel, A.; Day, M.J. Obesity and Associated Comorbidities in People and Companion Animals: A One Health Perspective. J. Comp. Pathol. 2017, 156, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.C. Nutritional therapies to improve health: Lessons from companion animals. Conference on “Multidisciplinary approaches to nutritional problems”. Symposium on “Nutrition and health”. Proc. Nutr. Soc. 2009, 68, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Costa-Santos, K.; Damasceno, K.; Portela, R.D.; Santos, F.L.; Araujo, G.C.; Martins-Filho, E.F.; Silva, L.P.; Barral, T.D.; Santos, S.A.; Estrela-Lima, A. Lipid and metabolic profiles in female dogs with mammary carcinoma receiving dietary fish oil supplementation. BMC Vet. Res. 2019, 15, 401. [Google Scholar] [CrossRef] [PubMed]
- Tvarijonaviciute, A.; Ceron, J.J.; Holden, S.L.; Cuthbertson, D.J.; Biourge, V.; Morris, P.J.; German, A.J. Obesity-related metabolic dysfunction in dogs: A comparison with human metabolic syndrome. BMC Vet. Res. 2012, 8, 147. [Google Scholar] [CrossRef]
- Montoya-Alonso, J.A.; Bautista-Castano, I.; Pena, C.; Suarez, L.; Juste, M.C.; Tvarijonaviciute, A. Prevalence of Canine Obesity, Obesity-Related Metabolic Dysfunction, and Relationship with Owner Obesity in an Obesogenic Region of Spain. Front. Vet. Sci. 2017, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M. Comparative Aspects of Diabetes Mellitus in Dogs and Cats. Mol. Cell. Endocrinol. 2002, 197, 221–229. [Google Scholar] [CrossRef]
- Stachowiak, M.; Szczerbal, I.; Switonski, M. Genetics of Adiposity in Large Animal Models for Human Obesity-Studies on Pigs and Dogs. Prog. Mol. Biol. Transl. Sci. 2016, 140, 233–270. [Google Scholar]
- German, A.J.; Blackwell, E.; Evans, M.; Westgarth, C. Overweight dogs are more likely to display undesirable behaviours: Results of a large online survey of dog owners in the UK. J. Nutr. Sci. 2017, 6, e14. [Google Scholar] [CrossRef]
- Raffan, E.; Smith, S.P.; O’Rahilly, S.; Wardle, J. Development, factor structure and application of the Dog Obesity Risk and Appetite (DORA) questionnaire. PeerJ 2015, 3, e1278. [Google Scholar] [CrossRef]
- Alegria-Moran, R.A.; Guzman-Pino, S.A.; Egana, J.I.; Munoz, C.; Figueroa, J. Food Preferences in Dogs: Effect of Dietary Composition and Intrinsic Variables on Diet Selection. Animals (Basel) 2019, 9, 372. [Google Scholar] [CrossRef]
- Gough, A.; Thomas, A.; O’Neill, D. Breed. Predispositions to Disease in Dogs and Cats, 3rd ed.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Farrell, L.L.; Schoenebeck, J.J.; Wiener, P.; Clements, D.N.; Summers, K.M. The challenges of pedigree dog health: Approaches to combating inherited disease. Canine Genet. Epidemiol. 2015, 2, 1–14. [Google Scholar] [CrossRef]
- Wilbe, M.; Jokinen, P.; Truve, K.; Seppala, E.H.; Karlsson, E.K.; Biagi, T.; Hughes, A.; Bannasch, D.; Andersson, G.; Hansson-Hamlin, H.; et al. Genome-wide association mapping identifies multiple loci for a canine SLE-related disease complex. Nat. Genet. 2010, 42, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Switonski, M.; Mankowska, M. Dog obesity—The need for identifying predisposing genetic markers. Res. Vet. Sci. 2013, 95, 831–836. [Google Scholar] [CrossRef]
- Mankowska, M.; Krzeminska, P.; Graczyk, M.; Switonski, M. Confirmation that a deletion in the POMC gene is associated with body weight of Labrador Retriever dogs. Res. Vet. Sci. 2017, 112, 116–118. [Google Scholar] [CrossRef]
- Chandler, M. New thoughts about obesity. Companion Anim. 2018, 23, 686–695. [Google Scholar] [CrossRef]
- Crane, S.W. Occurrence and Management of Obesity in Companion Animals. J. Small Anim. Pract. 1991, 31, 275–282. [Google Scholar] [CrossRef]
- Plassais, J.; Rimbault, M.; Williams, F.J.; Davis, B.W.; Schoenebeck, J.J.; Ostrander, E.A. Analysis of large versus small dogs reveals three genes on the canine X chromosome associated with body weight, muscling and back fat thickness. PLoS Genet. 2017, 13, e1006661. [Google Scholar] [CrossRef]
- Mawby, D.I.; Bartges, J.W.; d’Avignon, A.; Laflamme, D.P.; Moyers, T.D.; Cottrell, T. Comparison of various methods for estimating body fat in dogs. J. Am. Anim. Hosp. Assoc. 2004, 40, 109–114. [Google Scholar] [CrossRef]
- Laflamme, D.R. Development and validation of a body condition score system for dogs.: A clinical tool. Canine Pract. 1997, 22, 10–15. [Google Scholar]
- German, A.J.; Holden, S.L.; Moxham, G.L.; Holmes, K.L.; Hackett, R.M.; Rawlings, J.M. A simple, reliable tool for owners to assess the body condition of their dog or cat. J. Nutr. 2006, 136, 2031S–2033S. [Google Scholar] [CrossRef] [PubMed]
- Davison, L.J.; Holder, A.; Catchpole, B.; O’Callaghan, C.A. The Canine POMC Gene, Obesity in Labrador Retrievers and Susceptibility to Diabetes Mellitus. J. Vet. Intern. Med. 2017, 31, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, N.O.; Albuquerque, A.L.H.; Basso, R.M.; Trecenti, A.S.; Albertino, L.G.; Melchert, A.; Borges, A.S.; Oliveira-Filho, J.P. Canine POMC deletion (P187fs) allele frequency in Labrador Retrievers in Brazil. Pesqui. Vet. Bras. 2019, 39, 909–914. [Google Scholar] [CrossRef]
- Lee, Y.S.; Challis, B.G.; Thompson, D.A.; Yeo, G.S.; Keogh, J.M.; Madonna, M.E.; Wraight, V.; Sims, M.; Vatin, V.; Meyre, D.; et al. A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab. 2006, 3, 135–140. [Google Scholar] [CrossRef]
- Challis, B.G.; Pritchard, L.E.; Creemers, J.W.; Delplanque, J.; Keogh, J.M.; Luan, J.; Wareham, N.J.; Yeo, G.S.; Bhattacharyya, S.; Froguel, P.; et al. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum. Mol. Genet. 2002, 11, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, C.E.; Keogh, J.M.; Greenfield, J.R.; Henning, E.; van der Klaauw, A.A.; Blackwood, A.; O’Rahilly, S.; Roelfsema, F.; Camacho-Hubner, C.; Pijl, H.; et al. Obesity due to melanocortin 4 receptor (MC4R) deficiency is associated with increased linear growth and final height, fasting hyperinsulinemia, and incompletely suppressed growth hormone secretion. J. Clin. Endocrinol. Metab. 2011, 96, E181–E188. [Google Scholar] [CrossRef]
- Yan, J.; Tao, Y.X. Pharmacological characterization of canine melancortin-4 receptor and its natural variant V213F. Domest. Anim. Endocrinol. 2011, 41, 91–97. [Google Scholar] [CrossRef]
- Skorczyk, A.; Stachowiak, M.; Szczerbal, I.; Klukowska-Roetzler, J.; Schelling, C.; Dolf, G.; Switonski, M. Polymorphism and chromosomal location of the MC4R (melanocortin-4 receptor) gene in the dog and red fox. Gene 2007, 392, 247–252. [Google Scholar] [CrossRef]
- van den Berg, L.; van den Berg, S.M.; Martens, E.E.; Hazewinkel, H.A.; Dijkshoorn, N.A.; Delemarre-van de Waal, H.A.; Heutink, P.; Leegwater, P.A.; Heuven, H.C. Analysis of variation in the melanocortin-4 receptor gene (mc4r) in Golden Retriever dogs. Anim. Genet. 2010, 41, 557. [Google Scholar] [CrossRef]
- Zeng, R.; Zhang, Y.; Du, P. SNPs of melanocortin 4 receptor (MC4R) associated with body weight in Beagle dogs. Exp. Anim. 2014, 63, 73–78. [Google Scholar] [CrossRef]
- Mankowska, M.; Nowacka-Woszuk, J.; Graczyk, A.; Ciazynska, P.; Stachowiak, M.; Switonski, M. Polymorphism and methylation of the MC4R gene in obese and non-obese dogs. Mol. Biol. Rep. 2017, 44, 333–339. [Google Scholar] [CrossRef]
- Dubern, B.; Clement, K.; Pelloux, V.; Froguel, P.; Girardet, J.P.; Guy-Grand, B.; Tounian, P. Mutational analysis of melanocortin-4 receptor, agouti-related protein, and alpha-melanocyte-stimulating hormone genes in severely obese children. J. Pediatr. 2001, 139, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Grzes, M.; Szczerbal, I.; Fijak-Nowak, H.; Szydlowski, M.; Switonski, M. Two candidate genes (FTO and INSIG2) for fat accumulation in four canids: Chromosome mapping, gene polymorphisms and association studies of body and skin weight of red foxes. Cytogenet. Genome Res. 2011, 135, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Grzemski, A.; Stachowiak, M.; Flisikowski, K.; Mankowska, M.; Krzeminska, P.; Gogulski, M.; Aleksiewicz, R.; Szydlowski, M.; Switonski, M.; Nowacka-Woszuk, J. FTO and IRX3 genes are not promising markers for obesity in Labrador retriever dogs. Ann. Anim. Sci. 2019, 19, 343–357. [Google Scholar] [CrossRef]
- Skorczyk, A.; Flisikowski, K.; Szydlowski, M.; Cieslak, J.; Fries, R.; Switonski, M. Association of MC3R gene polymorphisms with body weight in the red fox and comparative gene organization in four canids. Anim. Genet. 2011, 42, 104–107. [Google Scholar] [CrossRef]
- Lee, B.; Koo, J.; Yun Jun, J.; Gavrilova, O.; Lee, Y.; Seo, A.Y.; Taylor-Douglas, D.C.; Adler-Wailes, D.C.; Chen, F.; Gardner, R.; et al. A mouse model for a partially inactive obesity-associated human MC3R variant. Nat. Commun. 2016, 7, 10522. [Google Scholar] [CrossRef]
- Tao, Y.X. Mutations in the melanocortin-3 receptor (MC3R) gene: Impact on human obesity or adiposity. Curr. Opin. Investig. Drugs 2010, 11, 1092–1096. [Google Scholar]
- Ghamari-Langroudi, M.; Cakir, I.; Lippert, R.N.; Sweeney, P.; Litt, M.J.; Ellacott, K.L.J.; Cone, R.D. Regulation of energy rheostasis by the melanocortin-3 receptor. Sci. Adv. 2018, 4, eaat0866. [Google Scholar] [CrossRef]
- Girardet, C.; Begriche, K.; Ptitsyn, A.; Koza, R.A.; Butler, A.A. Unravelling the mysterious roles of melanocortin-3 receptors in metabolic homeostasis and obesity using mouse genetics. Int. J. Obes. Suppl. 2014, 4, S37–S44. [Google Scholar] [CrossRef]
- Herbert, A.; Gerry, N.P.; McQueen, M.B.; Heid, I.M.; Pfeufer, A.; Illig, T.; Wichmann, H.E.; Meitinger, T.; Hunter, D.; Hu, F.B.; et al. A common genetic variant is associated with adult and childhood obesity. Science 2006, 312, 279–283. [Google Scholar] [CrossRef]
- Hinney, A.; Hebebrand, J. Polygenic obesity in humans. Obes. Facts 2008, 1, 35–42. [Google Scholar] [CrossRef]
- Hudson, B.D.; Shimpukade, B.; Mackenzie, A.E.; Butcher, A.J.; Pediani, J.D.; Christiansen, E.; Heathcote, H.; Tobin, A.B.; Ulven, T.; Milligan, G. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol. Pharmacol. 2013, 84, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.M.; Dhayal, S.; Brocklehurst, K.J.; Lenaghan, C.; Sorhede Winzell, M.; Hammar, M.; Xu, X.; Smith, D.M.; Morgan, N.G. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia 2014, 57, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, A.; Hirasawa, A.; Poulain-Godefroy, O.; Bonnefond, A.; Hara, T.; Yengo, L.; Kimura, I.; Leloire, A.; Liu, N.; Iida, K.; et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 2012, 483, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, M.; Gin, A.; Onozawa, E.; Daimon, M.; Yamada, H.; Oda, H.; Mori, A.; Momota, Y.; Azakami, D.; Yamamoto, I.; et al. Genetic variants of the unsaturated fatty acid receptor GPR120 relating to obesity in dogs. J. Vet. Med. Sci. 2015, 77, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Kim, S. Genetic manipulations of PPARs: Effects on obesity and metabolic disease. PPAR Res. 2007, 2007, 12781. [Google Scholar] [CrossRef]
- Vidal-Puig, A.; Jimenez-Linan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef]
- Nishii, N.; Takasu, M.; Soe, O.K.; Maeda, S.; Ohba, Y.; Inoue-Murayama, M.; Kitagawa, H. Cloning, expression and investigation for polymorphisms of canine peroxisome proliferator-activated receptors. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2007, 147, 690–697. [Google Scholar] [CrossRef]
- Mankowska, M.; Stachowiak, M.; Graczyk, A.; Ciazynska, P.; Gogulski, M.; Nizanski, W.; Switonski, M. Sequence analysis of three canine adipokine genes revealed an association between TNF polymorphisms and obesity in Labrador dogs. Anim. Genet. 2016, 47, 245–249. [Google Scholar] [CrossRef]
- Yu, Z.; Han, S.; Cao, X.; Zhu, C.; Wang, X.; Guo, X. Genetic polymorphisms in adipokine genes and the risk of obesity: A systematic review and meta-analysis. Obesity (Silver Spring) 2012, 20, 396–406. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Vandendriessche, V.L.; Picavet, P.; Hesta, M. First detailed nutritional survey in a referral companion animal population. J. Anim. Physiol. Anim. Nutr. (Berl.) 2017, 101 (Suppl. S1), S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Courcier, E.A.; O’Higgins, R.; Mellor, D.J.; Yam, P.S. Prevalence and risk factors for feline obesity in a first opinion practice in Glasgow, Scotland. J. Feline Med. Surg. 2010, 12, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, J.M.; Donoghue, S.; Saidla, J.; Wills, J. Overweight cats: Prevalence and risk factors. Int. J. Obes. Relat. Metab. Disord. 1994, 18 (Suppl. S1), S22–S28. [Google Scholar] [PubMed]
- Courcier, E.A.; Mellor, D.J.; Pendlebury, E.; Evans, C.; Yam, P.S. An investigation into the epidemiology of feline obesity in Great Britain: Results of a cross-sectional study of 47 companion animal practises. Vet. Rec. 2012, 171, 560. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.; Cave, N.J.; Vallee, E. Owner and Cat-Related Risk Factors for Feline Overweight or Obesity. Front. Vet. Sci. 2019, 6, 266. [Google Scholar] [CrossRef]
- Tarkosova, D.; Story, M.M.; Rand, J.S.; Svoboda, M. Feline obesity–prevalence, risk factors, pathogenesis, associated conditions and assessment: A review. Vet. Med. 2016, 61, 295–307. [Google Scholar] [CrossRef]
- Scarlett, J.M.; Donoghue, S. Associations between body condition and disease in cats. J. Am. Vet. Med. Assoc. 1998, 212, 1725–1731. [Google Scholar]
- Kocabağlı, N.; Kutay, H.C.; Dokuzeylül, B.; Süer, İ.N.E.; Apt, M. The Analysis of Computer Data regarding Obesity and Associated Diseases in Cats Examined at Private Veterinary Practices. Acta Sci. Vet. 2017, 45, 5. [Google Scholar] [CrossRef]
- Center, S.A. Feline hepatic lipidosis. Vet. Clin. N. Am. Small Anim. Pract. 2005, 35, 225–269. [Google Scholar] [CrossRef]
- Hoenig, M.; Thomaseth, K.; Waldron, M.; Ferguson, D.C. Insulin sensitivity, fat distribution, and adipocytokine response to different diets in lean and obese cats before and after weight loss. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R227–R234. [Google Scholar] [CrossRef]
- Raffan, E. The big problem: Battling companion animal obesity. Vet. Rec. 2013, 173, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, H.S.; Galac, S.; Buijtels, J.J.; Meij, B.P. Endocrine diseases in animals. Horm. Res. 2009, 71 (Suppl. S1), S144–S147. [Google Scholar] [CrossRef] [PubMed]
- Van de Velde, H.; Janssens, G.P.; de Rooster, H.; Polis, I.; Peters, I.; Ducatelle, R.; Nguyen, P.; Buyse, J.; Rochus, K.; Xu, J.; et al. The cat as a model for human obesity: Insights into depot-specific inflammation associated with feline obesity. Br. J. Nutr. 2013, 110, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Kley, S.; Le, N.A.; Waldron, M.; Hoenig, M. Dyslipidemia in obese cats. Domest. Anim. Endocrinol. 2008, 35, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M. The cat as a model for human obesity and diabetes. J. Diabetes Sci. Technol. 2012, 6, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Rand, J.S.; Fleeman, L.M.; Farrow, H.A.; Appleton, D.J.; Lederer, R. Canine and feline diabetes mellitus: Nature or nurture? J. Nutr. 2004, 134, 2072S–2080S. [Google Scholar] [CrossRef]
- Osto, M.; Lutz, T.A. Translational value of animal models of obesity-Focus on dogs and cats. Eur. J. Pharmacol. 2015, 759, 240–252. [Google Scholar] [CrossRef]
- Osto, M.; Zini, E.; Reusch, C.E.; Lutz, T.A. Diabetes from humans to cats. Gen. Comp. Endocrinol. 2013, 182, 48–53. [Google Scholar] [CrossRef]
- Zini, E.; Osto, M.; Franchini, M.; Guscetti, F.; Donath, M.Y.; Perren, A.; Heller, R.S.; Linscheid, P.; Bouwman, M.; Ackermann, M.; et al. Hyperglycaemia but not hyperlipidaemia causes beta cell dysfunction and beta cell loss in the domestic cat. Diabetologia 2009, 52, 336–346. [Google Scholar] [CrossRef]
- Clark, M.; Hoenig, M. Metabolic Effects of Obesity and Its Interaction with Endocrine Diseases. Vet. Clin. N. Am. Small Anim. Pract. 2016, 46, 797–815. [Google Scholar] [CrossRef]
- Häring, T.; Haase, B.; Zini, E.; Hartnack, S.; Uebelhart, D.; Gaudenz, D.; Wichert, B.A. Overweight and impaired insulin sensitivity present in growing cats. J. Anim. Physiol. Anim. Nutr. (Berl.) 2013, 97, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Center, S.A.; Crawford, M.A.; Guida, L.; Erb, H.N.; King, J. A retrospective study of 77 cats with severe hepatic lipidosis: 1975-1990. J. Vet. Intern. Med. 1993, 7, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Verbrugghe, A.; Bakovic, M. Peculiarities of one-carbon metabolism in the strict carnivorous cat and the role in feline hepatic lipidosis. Nutrients 2013, 5, 2811–2835. [Google Scholar] [CrossRef] [PubMed]
- Corbee, R.J. Obesity in show cats. J. Anim. Physiol. Anim. Nutr. (Berl.) 2014, 98, 1075–1080. [Google Scholar] [CrossRef]
- Ohlund, M.; Palmgren, M.; Holst, B.S. Overweight in adult cats: A cross-sectional study. Acta Vet. Scand. 2018, 60, 5. [Google Scholar] [CrossRef]
- Colliard, L.; Paragon, B.M.; Lemuet, B.; Benet, J.J.; Blanchard, G. Prevalence and risk factors of obesity in an urban population of healthy cats. J. Feline Med. Surg. 2009, 11, 135–140. [Google Scholar] [CrossRef]
- Alhaddad, H.; Khan, R.; Grahn, R.A.; Gandolfi, B.; Mullikin, J.C.; Cole, S.A.; Gruffydd-Jones, T.J.; Haggstrom, J.; Lohi, H.; Longeri, M.; et al. Extent of linkage disequilibrium in the domestic cat, Felis silvestris catus, and its breeds. PLoS ONE 2013, 8, e53537. [Google Scholar] [CrossRef]
- Zhang, W.; Schoenebeck, J.J. The ninth life of the cat reference genome, Felis_catus. PLoS Genet. 2020, 16, e1009045. [Google Scholar] [CrossRef]
- Forcada, Y.; Holder, A.; Church, D.B.; Catchpole, B. A polymorphism in the melanocortin 4 receptor gene (MC4R:c.92C>T) is associated with diabetes mellitus in overweight domestic shorthaired cats. J. Vet. Intern. Med. 2014, 28, 458–464. [Google Scholar] [CrossRef]
- Häring, T.; Wichert, B.; Dolf, G.; Haase, B. Segregation analysis of overweight body condition in an experimental cat population. J. Hered. 2011, 102 (Suppl. S1), S28–S31. [Google Scholar]
- Wichert, B.; Trossen, J.; Uebelhart, D.; Wanner, M.; Hartnack, S. Energy requirement and food intake behaviour in young adult intact male cats with and without predisposition to overweight. Sci. World J. 2012, 2012, 509854. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Speakman, J.R.; Levitsky, D.A.; Allison, D.B.; Bray, M.S.; de Castro, J.M.; Clegg, D.J.; Clapham, J.C.; Dulloo, A.G.; Gruer, L.; Haw, S.; et al. Set points, settling points and some alternative models: Theoretical options to understand how genes and environments combine to regulate body adiposity. Dis. Model. Mech. 2011, 4, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Ghielmetti, V.; Wichert, B.; Ruegg, S.; Frey, D.; Liesegang, A. Food intake and energy expenditure in growing cats with and without a predisposition to overweight. J. Anim. Physiol. Anim. Nutr. (Berl.) 2018, 102, 1401–1410. [Google Scholar] [CrossRef]
- Keller, C.; Liesegang, A.; Frey, D.; Wichert, B. Metabolic response to three different diets in lean cats and cats predisposed to overweight. BMC Vet. Res. 2017, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Wichert, B.; Häring, T.; Dolf, G.; Trossen, J.; Haase, B.; Szymecko, R.; Iben, C.; Burlikowska, K.; Sitkowska, B. Feline Bodyweight: Genetic Aspects of Food Intake. In Proceedings of the 16th Congress of the European Society of Veterinary and Comparative Nutrition, Bydgoszcz, Poland, 13–15 September 2012; European Society of Veterinary & Comparative Nutrition: Zürich, Switzerland, 2012; Volume 31. [Google Scholar]
- Zobel, D.P.; Andreasen, C.H.; Grarup, N.; Eiberg, H.; Sorensen, T.I.; Sandbaek, A.; Lauritzen, T.; Borch-Johnsen, K.; Jorgensen, T.; Pedersen, O.; et al. Variants near MC4R are associated with obesity and influence obesity-related quantitative traits in a population of middle-aged people: Studies of 14,940 Danes. Diabetes 2009, 58, 757–764. [Google Scholar] [CrossRef]
- Cauchi, S.; Stutzmann, F.; Cavalcanti-Proenca, C.; Durand, E.; Pouta, A.; Hartikainen, A.L.; Marre, M.; Vol, S.; Tammelin, T.; Laitinen, J.; et al. Combined effects of MC4R and FTO common genetic variants on obesity in European general populations. J. Mol. Med. (Berl.) 2009, 87, 537–546. [Google Scholar] [CrossRef]
- Forcada, Y.; Boursnell, M.; Catchpole, B.; Church, D.B. A Genome-Wide Association Study Identifies Novel Candidate Genes for Susceptibility to Diabetes Mellitus in DSH Cats. In Proceedings of the Conference Proceedings: 25th ECVIM-CA Congress, Lisbon, Portugal, 10–12 September 2015. [Google Scholar]
- Forcada, Y.; Boursnell, M.; Catchpole, B.; Church, D.B. A Genome-Wide Association Study Identifies Novel Candidate Genes for the Susceptibility to Diabetes Mellitus in DSH Cats. In Proceedings of the Conference Proceedings: ACVIM, Am College Vet Internal Med Forum, Denver, CO, USA, 9–11 June 2016. [Google Scholar]
- Hazuchova, H.; Wallace, M.; Church, D.B.; Catchpole, B.; Forcada, Y. Analysis of GWAS Data in Domestic Shorthair and Burmese Cats Identifies Diabetes-associated Loci Near the DPP9 and Within the DPP10 Gene. In Proceedings of the Conference Proceedings: 29th ECVIM-CA Congress, European Coll Vet Int Med, Milano, Italy, 19–21 September 2019. [Google Scholar]
- Rendle, D.; McGregor-Argo, C.; Bowen, M.; Carslake, H.; German, A.; Harris, P.; Knowles, E.; Menzies-Gow, N.; Morgan, R. Equine obesity: Current perspectives. UK Vet. Equine 2018, 2, 1–19. [Google Scholar] [CrossRef]
- McCue, M.E.; Geor, R.J.; Schultz, N. Equine metabolic syndrome: A complex disease influenced by genetics and the environment. J. Equine Vet. Sci. 2015, 35, 367–375. [Google Scholar] [CrossRef]
- Thatcher, C.D.; Pleasant, R.S.; Geor, R.J.; Elvinger, F. Prevalence of overconditioning in mature horses in southwest Virginia during the summer. J. Vet. Intern. Med. 2012, 26, 1413–1418. [Google Scholar] [CrossRef]
- Stephenson, H.M.; Green, M.J.; Freeman, S.L. Prevalence of obesity in a population of horses in the UK. Vet. Rec. 2011, 168, 131. [Google Scholar] [CrossRef]
- Johnson, P.J. The equine metabolic syndrome peripheral Cushing’s syndrome. Vet. Clin. N. Am. Equine Pract. 2002, 18, 271–293. [Google Scholar] [CrossRef]
- Durham, A.E.; Frank, N.; McGowan, C.M.; Menzies-Gow, N.J.; Roelfsema, E.; Vervuert, I.; Feige, K.; Fey, K. ECEIM consensus statement on equine metabolic syndrome. J. Vet. Intern. Med. 2019, 33, 335–349. [Google Scholar] [CrossRef] [PubMed]
- de Laat, M.A.; McGree, J.M.; Sillence, M.N. Equine hyperinsulinemia: Investigation of the enteroinsular axis during insulin dysregulation. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E61–E72. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.J.; Bamford, N.J.; Harris, P.A.; Bailey, S.R. Prevalence of obesity and owners’ perceptions of body condition in pleasure horses and ponies in south-eastern Australia. Aust. Vet. J. 2016, 94, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Giles, S.L.; Rands, S.A.; Nicol, C.J.; Harris, P.A. Obesity prevalence and associated risk factors in outdoor living domestic horses and ponies. PeerJ 2014, 2, e299. [Google Scholar] [CrossRef]
- Morrison, P.K.; Harris, P.A.; Maltin, C.A.; Grove-White, D.; Barfoot, C.F.; Argo, C.M. Perceptions of obesity and management practices in a UK population of leisure-horse owners and managers. J. Equine Vet. Sci. 2017, 53, 19–29. [Google Scholar] [CrossRef]
- Jensen, R.B.; Danielsen, S.H.; Tauson, A.H. Body condition score, morphometric measurements and estimation of body weight in mature Icelandic horses in Denmark. Acta Vet. Scand. 2016, 58, 59. [Google Scholar] [CrossRef]
- Harker, I.J.; Harris, P.A.; Barfoot, C.F. The body condition score of leisure horses competing at an unaffiliated championship in the UK. J. Equine Vet. Sci. 2011, 5, 253–254. [Google Scholar] [CrossRef]
- Sánchez-Guerrero, M.J.; Ramos, J.; Valdés, M.; Valera, M. Prevalence, Environmental Risk Factors and Heritability of Body Condition in Pura Raza Español Horses. Livest. Sci. 2019, 230, 103851. [Google Scholar] [CrossRef]
- Bamford, N.J.; Potter, S.J.; Harris, P.A.; Bailey, S.R. Breed differences in insulin sensitivity and insulinemic responses to oral glucose in horses and ponies of moderate body condition score. Domest. Anim. Endocrinol. 2014, 47, 101–107. [Google Scholar] [CrossRef]
- Freestone, J.F.; Shoemaker, K.; Bessin, R.; Wolfsheimer, J.K. Insulin and glucose response following oral glucose administration in well-conditioned ponies. Equine Vet. J. Suppl. 1992, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Jeffcott, L.B.; Field, J.R.; McLean, J.G.; O’Dea, K. Glucose tolerance and insulin sensitivity in ponies and Standardbred horses. Equine Vet. J. 1986, 18, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Robie, S.M.; Janson, C.H.; Smith, S.C.; O’Connor, J.T., Jr. Equine serum lipids: Serum lipids and glucose in Morgan and Thoroughbred horses and Shetland ponies. Am. J. Vet. Res. 1975, 36, 1705–1708. [Google Scholar] [PubMed]
- Norton, E.M.; Schultz, N.E.; Rendahl, A.K.; Mcfarlane, D.; Geor, R.J.; Mickelson, J.R.; McCue, M.E. Heritability of Metabolic Traits Associated with Equine Metabolic Syndrome in Welsh Ponies and Morgan Horses. Equine Vet. J. 2019, 51, 475–480. [Google Scholar] [CrossRef]
- Treiber, K.H.; Kronfeld, D.S.; Hess, T.M.; Byrd, B.M.; Splan, R.K.; Staniar, W.B. Evaluation of genetic and metabolic predispositions and nutritional risk factors for pasture-associated laminitis in ponies. J. Am. Vet. Med. Assoc. 2006, 228, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.L.; Holl, H.M.; Streeter, C.; Posbergh, C.; Schanbacher, B.J.; Place, N.J.; Mallicote, M.F.; Long, M.T.; Brooks, S.A. Genomewide association study reveals a risk locus for equine metabolic syndrome in the Arabian horse. J. Anim. Sci. 2017, 95, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Cash, C.M.; Fitzgerald, D.M.; Spence, R.J.; de Laat, M.A. Preliminary analysis of the FAM174A gene suggests it lacks a strong association with equine metabolic syndrome in ponies. Domest. Anim. Endocrinol. 2020, 72, 106439. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.M.; Norton, E.M.; Rendahl, A.K.; Schultz, N.E.; McFarlane, D.; Geor, R.J.; Mickelson, J.R.; McCue, M.E. Assessment of the FAM174A 11G allele as a risk allele for equine metabolic syndrome. Anim. Genet. 2020, 51, 607–610. [Google Scholar] [CrossRef]
- Norton, E.; Schultz, N.; Geor, R.; McFarlane, D.; Mickelson, J.; McCue, M. Genome-Wide Association Analyses of Equine Metabolic Syndrome Phenotypes in Welsh Ponies and Morgan Horses. Genes (Basel) 2019, 10, 893. [Google Scholar] [CrossRef]
- Norton, E.M.; Avila, F.; Schultz, N.E.; Mickelson, J.R.; Geor, R.J.; McCue, M.E. Evaluation of an HMGA2 variant for pleiotropic effects on height and metabolic traits in ponies. J. Vet. Intern. Med. 2019, 33, 942–952. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.J.; Castelhano, M.G.; Oliveira, K.C.; Corey, E.; Balkman, C.; Baxter, T.L.; Casal, M.L.; Center, S.A.; Fang, M.; Garrison, S.J.; et al. Complex disease and phenotype mapping in the domestic dog. Nat. Commun. 2016, 7, 10460. [Google Scholar] [CrossRef] [PubMed]
- Dodman, N.H.; Karlsson, E.K.; Moon-Fanelli, A.; Galdzicka, M.; Perloski, M.; Shuster, L.; Lindblad-Toh, K.; Ginns, E.I. A canine chromosome 7 locus confers compulsive disorder susceptibility. Mol. Psychiatry 2010, 15, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Hoeppner, M.P.; Lundquist, A.; Pirun, M.; Meadows, J.R.; Zamani, N.; Johnson, J.; Sundstrom, G.; Cook, A.; FitzGerald, M.G.; Swofford, R.; et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS ONE 2014, 9, e91172. [Google Scholar] [CrossRef]
- Halo, J.; Pendelton, A.L.; Shen, F.; Doucet, A.J.; Derrien, T.; Hitte, C.; Kirby, L.E.; Myers, B.; Sliwerska, E.; Emery, S.; et al. Preprint—Long-read assembly of a Great Dane genome highlights the contribution of GC-rich sequence and mobile elements to canine genomes. bioRxiv 2020. preprints. [Google Scholar] [CrossRef]
- Bannasch, D.; Young, A.; Myers, J.; Truve, K.; Dickinson, P.; Gregg, J.; Davis, R.; Bongcam-Rudloff, E.; Webster, M.T.; Lindblad-Toh, K.; et al. Localization of canine brachycephaly using an across breed mapping approach. PLoS ONE 2010, 5, e9632. [Google Scholar] [CrossRef]
- Friedenberg, S.G.; Meurs, K.M. Genotype imputation in the domestic dog. Mamm. Genome 2016, 27, 485–494. [Google Scholar] [CrossRef]
- Hayward, J.J.; White, M.E.; Boyle, M.; Shannon, L.M.; Casal, M.L.; Castelhano, M.G.; Center, S.A.; Meyers-Wallen, V.N.; Simpson, K.W.; Sutter, N.B.; et al. Imputation of canine genotype array data using 365 whole-genome sequences improves power of genome-wide association studies. PLoS Genet. 2019, 15, e1008003. [Google Scholar] [CrossRef]
- Gandolfi, B.; Alhaddad, H.; Abdi, M.; Bach, L.H.; Creighton, E.K.; Davis, B.W.; Decker, J.E.; Dodman, N.H.; Ginns, E.I.; Grahn, J.C.; et al. Applications and efficiencies of the first cat 63K DNA array. Sci. Rep. 2018, 8, 7024. [Google Scholar] [CrossRef]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O’Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Petersen, J.L.; et al. Preprint—EquCab3, an updated reference genome for the domestic horse. bioRxiv 2018, 306928. [Google Scholar] [CrossRef]
- McCue, M.E.; Bannasch, D.L.; Petersen, J.L.; Gurr, J.; Bailey, E.; Binns, M.M.; Distl, O.; Guerin, G.; Hasegawa, T.; Hill, E.W.; et al. A high density SNP array for the domestic horse and extant Perissodactyla: Utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012, 8, e1002451. [Google Scholar] [CrossRef]
- Schaefer, R.J.; Schubert, M.; Bailey, E.; Bannasch, D.L.; Barrey, E.; Bar-Gal, G.K.; Brem, G.; Brooks, S.A.; Distl, O.; Fries, R.; et al. Developing a 670k genotyping array to tag ~2M SNPs across 24 horse breeds. BMC Genom. 2017, 18, 565. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, R.J.; McCue, M.E. Equine Genotyping Arrays. Vet. Clin. N. Am. Equine Pract. 2020, 36, 183–193. [Google Scholar] [CrossRef]
- Chassier, M.; Barrey, E.; Robert, C.; Duluard, A.; Danvy, S.; Ricard, A. Genotype imputation accuracy in multiple equine breeds from medium- to high-density genotypes. J. Anim. Breed. Genet. 2018, 135, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Samaha, G.; Beatty, J.; Wade, C.M.; Haase, B. The Burmese cat as a genetic model of type 2 diabetes in humans. Anim. Genet. 2019, 50, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Brito-Casillas, Y.; Melian, C.; Wagner, A.M. Study of the pathogenesis and treatment of diabetes mellitus through animal models. Endocrinol. Nutr. 2016, 63, 345–353. [Google Scholar] [CrossRef]
- Srinivasan, K.; Ramarao, P. Animal models in type 2 diabetes research: An overview. Indian J. Med. Res. 2007, 125, 451–472. [Google Scholar]
- Rimbault, M.; Ostrander, E.A. So many doggone traits: Mapping genetics of multiple phenotypes in the domestic dog. Hum. Mol. Genet. 2012, 21, R52–R57. [Google Scholar] [CrossRef]
- Shearin, A.L.; Ostrander, E.A. Leading the way: Canine models of genomics and disease. Dis. Model. Mech. 2010, 3, 27–34. [Google Scholar] [CrossRef]
- Switonski, M. Dog as a model in studies on human hereditary diseases and their gene therapy. Reprod. Biol. 2014, 14, 44–50. [Google Scholar] [CrossRef]
- Momozawa, Y.; Merveille, A.C.; Battaille, G.; Wiberg, M.; Koch, J.; Willesen, J.L.; Proschowsky, H.F.; Gouni, V.; Chetboul, V.; Tiret, L.; et al. Genome wide association study of 40 clinical measurements in eight dog breeds. Sci. Rep. 2020, 10, 6520. [Google Scholar] [CrossRef]
- Sutter, N.B.; Eberle, M.A.; Parker, H.G.; Pullar, B.J.; Kirkness, E.F.; Kruglyak, L.; Ostrander, E.A. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. 2004, 14, 2388–2396. [Google Scholar] [CrossRef]
- Marsden, C.D.; Ortega-Del Vecchyo, D.; O’Brien, D.P.; Taylor, J.F.; Ramirez, O.; Vila, C.; Marques-Bonet, T.; Schnabel, R.D.; Wayne, R.K.; Lohmueller, K.E. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. USA 2016, 113, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Gurda, B.L.; Bradbury, A.M.; Vite, C.H. Canine and Feline Models of Human Genetic Diseases and Their Contributions to Advancing Clinical Therapies. Yale J. Biol. Med. 2017, 90, 417–431. [Google Scholar] [PubMed]
- Oh, A.; Pearce, J.W.; Gandolfi, B.; Ceighton, E.K.; Suedmeyer, W.K.; Selig, M.; Bosiack, A.P.; Castaner, L.J.; Whiting, R.E.; Belknap, E.B.; et al. Early-Onset Progressive Retinal Atrophy Associated with an IQCB1 Variant in African Black-Footed Cats (Felis nigripes). Sci. Rep. 2017, 7, 43918. [Google Scholar] [CrossRef] [PubMed]
- Lonker, N.S.; Fechner, K.; Wahed, A.A.E. Horses as a Crucial Part of One Health. Vet. Sci. 2020, 7, 28. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallis, N.; Raffan, E. The Genetic Basis of Obesity and Related Metabolic Diseases in Humans and Companion Animals. Genes 2020, 11, 1378. https://doi.org/10.3390/genes11111378
Wallis N, Raffan E. The Genetic Basis of Obesity and Related Metabolic Diseases in Humans and Companion Animals. Genes. 2020; 11(11):1378. https://doi.org/10.3390/genes11111378
Chicago/Turabian StyleWallis, Natalie, and Eleanor Raffan. 2020. "The Genetic Basis of Obesity and Related Metabolic Diseases in Humans and Companion Animals" Genes 11, no. 11: 1378. https://doi.org/10.3390/genes11111378
APA StyleWallis, N., & Raffan, E. (2020). The Genetic Basis of Obesity and Related Metabolic Diseases in Humans and Companion Animals. Genes, 11(11), 1378. https://doi.org/10.3390/genes11111378