Segregation Analysis of Rare NRP1 and NRP2 Variants in Families with Lymphedema

 ,
,  , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Evaluation
2.2. Genetic Analysis
2.3. In-Silico Analysis
3. Results
3.1. Clinical and Genetic Evaluation
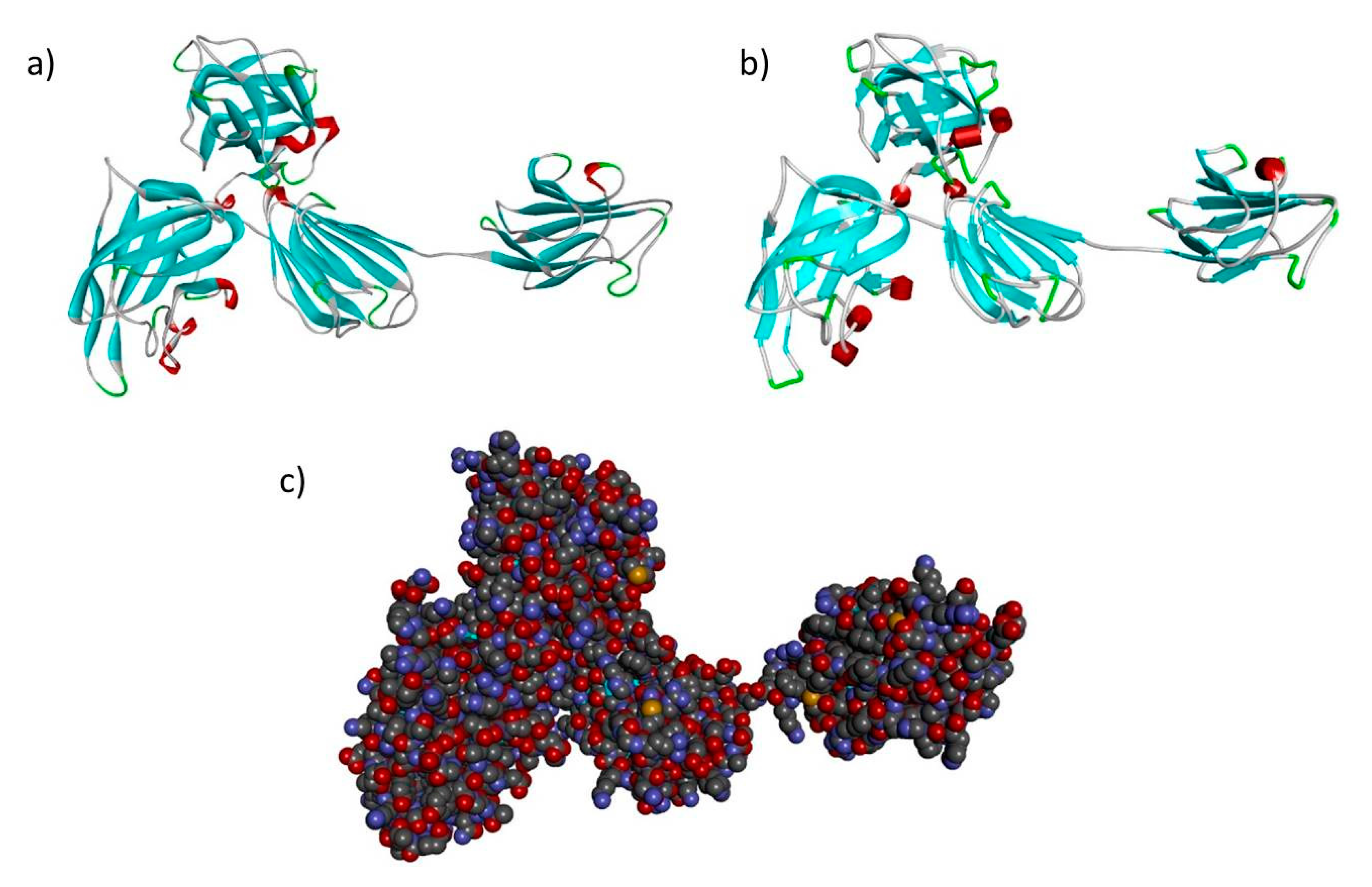
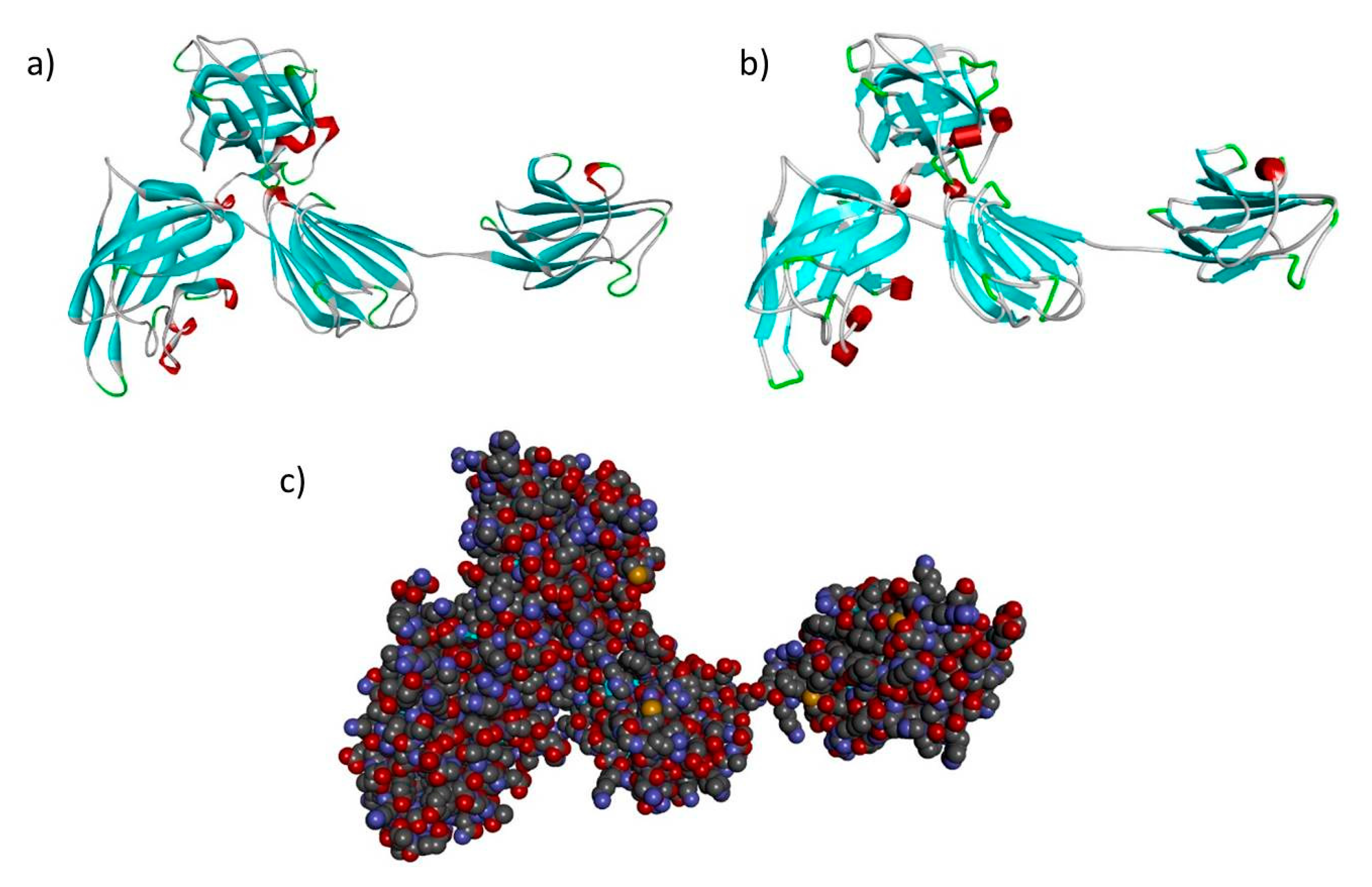
3.2. In Silico Analysis, Template Selection and Building
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next-Generation Sequencing |
| NRP1/2 | Neuropilin 1/2 |
| LECs | Lymphatic endothelial cells |
References
- Kolodkin, A.L.; Levengood, D.V.; Rowe, E.G.; Tai, Y.T.; Giger, R.J.; Ginty, D.D. Neuropilin is a semaphorin III receptor. Cell 1997, 90, 753–762. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Tessier-lavigne, M. Neuropilin Is a Receptor for the Axonal Chemorepellent Semaphorin III. Cell 1997, 90, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Giger, R.J.; Urquhart, E.R.; Gillespie, S.K.H.; Levengood, D.V.; Ginty, D.D.; Kolodkin, A.L. Neuropilin-2 is a receptor for semaphorin IV: Insight into the structural basis of receptor function and specificity. Neuron 1998, 21, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Gluzman-Poltorak, Z.; Cohen, T.; Herzog, Y.; Neufeld, G. Neuropilin-2 and neuropilin-1 are receptors for the 165-amino acid form of vascular endothelial growth factor (VEGF) and of placenta growth factor-2, but only neuropilin-2 functions as a receptor for the 145-amino acid form of VEGF. J. Biol. Chem. 2000, 275, 18040–18045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C.; Ruhrberg, C. Neuropilin signalling in vessels, neurons and tumours. Semin. Cell Dev. Biol. 2013, 24, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kärpänen, T.; Heckman, C.A.; Keskitalo, S.; Jeltsch, M.; Ollila, H.; Neufeld, G.; Tamagnone, L.; Alitalo, K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006, 20, 1462–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulpice, E.; Plouët, J.; Bergé, M.; Allanic, D.; Tobelem, G.; Merkulova-Rainon, T. Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood 2008, 111, 2036–2045. [Google Scholar] [CrossRef]
- Rizzolio, S.; Rabinowicz, N.; Rainero, E.; Lanzetti, L.; Serini, G.; Norman, J.; Neufeld, G.; Tamagnone, L. Neuropilin-1-dependent regulation of EGF-receptor Signaling. Cancer Res. 2012, 72, 5801–5811. [Google Scholar] [CrossRef] [Green Version]
- Castellani, V.; De Angelis, E.; Kenwrick, S.; Rougon, G. Cis and trans interactions of L1 with neuropilin-1 control axonal responses to semaphorin 3A. EMBO J. 2002, 21, 6348–6357. [Google Scholar] [CrossRef] [Green Version]
- Bagri, A.; Tessier-Lavigne, M.; Watts, R.J. Neuropilins in tumor biology. Clin. Cancer Res. 2009, 15, 1860–1864. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, G.; Kessler, O. The Neuropilins: Role and Function in Health and Disease; Springer International Publishing: Basel, Switzerland, 2017; ISBN 9783319488240. [Google Scholar]
- Bouvrée, K.; Brunet, I.; Del Toro, R.; Gordon, E.; Prahst, C.; Cristofaro, B.; Mathivet, T.; Xu, Y.; Soueid, J.; Fortuna, V.; et al. Semaphorin3A, Neuropilin-1, and PlexinA1 are required for lymphatic valve formation. Circ. Res. 2012, 111, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yuan, L.; Mak, J.; Pardanaud, L.; Caunt, M.; Kasman, I.; Larrivée, B.; Del Toro, R.; Suchting, S.; Medvinsky, A.; et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 2010, 188, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kitsukawa, T.; Bekku, Y.; Matsuda, Y.; Sanbo, M.; Yagi, T.; Fujisawa, H. A requirement for neuropilin-1 in embryonic vessel formation. Development 1999, 126, 4895–4902. [Google Scholar]
- Kitsukawa, T.; Shimizu, M.; Sanbo, M.; Hirata, T.; Taniguchi, M.; Bekku, Y.; Yagi, T.; Fujisawa, H. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron 1997, 19, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Kitsukawa, T.; Shimono, A.; Kawakami, A.; Kondoh, H.; Fujisawa, H. Overexpression of a membrane protein, neuropilin, in chimeric mice causes anomalies in the cardiovascular system, nervous system and limbs. Development 1995, 121, 4309–4318. [Google Scholar]
- Jurisic, G.; Maby-El Hajjami, H.; Karaman, S.; Ochsenbein, A.M.; Alitalo, A.; Siddiqui, S.S.; Ochoa Pereira, C.; Petrova, T.V.; Detmar, M. An Unexpected Role of Semaphorin3A–Neuropilin-1 Signaling in Lymphatic Vessel Maturation and Valve Formation. Circ. Res. 2012, 111, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Aspelund, A.; Alitalo, K. Lymphangiogenic factors, mechanisms, and applications. J. Clin. Investig. 2014, 124, 878–887. [Google Scholar] [CrossRef]
- Yuan, L.; Moyon, D.; Pardanaud, L.; Bréant, C.; Karkkainen, M.J.; Alitalo, K.; Eichmann, A. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 2002, 129, 4797–4806. [Google Scholar]
- Takashima, S.; Kitakaze, M.; Asakura, M.; Asanuma, H.; Sanada, S.; Tashiro, F.; Niwa, H.; Miyazaki, J.I.; Hirota, S.; Kitamura, Y.; et al. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 3657–3662. [Google Scholar] [CrossRef] [Green Version]
- Michelini, S.; Cardone, M.; Maltese, P.; Bruson, A.; Fiorentino, A.; Bertelli, M. Primary lymphedema and genetic implications. EuroBiotech J. 2017, 1, 144–146. [Google Scholar]
- Petrova, T.V.; Karpanen, T.; Norrmén, C.; Mellor, R.; Tamakoshi, T.; Finegold, D.; Ferrell, R.; Kerjaschki, D.; Mortimer, P.; Ylä-Herttuala, S.; et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 2004, 10, 974–981. [Google Scholar] [CrossRef]
- Michelini, S.; Ricci, M.; Veselenyiova, D.; Kenanoglu, S.; Kurti, D.; Baglivo, M.; Fiorentino, A.; Basha, S.H.; Priya, S.; Serrani, R.; et al. TIE1 as a candidate gene for lymphatic malformations with or without lymphedema. Int. J. Mol. Sci. 2020, 21, 6780. [Google Scholar] [CrossRef] [PubMed]
- Michelini, S.; Paolacci, S.; Manara, E.; Eretta, C.; Mattassi, R.; Lee, B.-B.; Bertelli, M. Genetic tests in lymphatic vascular malformations and lymphedema. J. Med. Genet. 2018, 55, 222–232. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, 162–173. [Google Scholar] [CrossRef]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulation. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Studio, B.D. Discovery Studio Modeling Environment [Internet]; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2016. [Google Scholar]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E.; The Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltese, P.E.; Michelini, S.; Ricci, M.; Maitz, S.; Fiorentino, A.; Serrani, R.; Lazzerotti, A.; Bruson, A.; Paolacci, S.; Benedetti, S.; et al. Increasing evidence of hereditary lymphedema caused by CELSR1 loss-of-function variants. Am. J. Med. Genet. 2019, 179, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Brouillard, P.; Boon, L.; Vikkula, M. Genetics of lymphatic anomalies. J. Clin. Investig. 2014, 124, 898–904. [Google Scholar] [CrossRef]
- Schwarz, Q.; Ruhrberg, C. Neuropilin, you gotta let me know: Should I stay or should I go? Cell Adhes. Migr. 2010, 4, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Connell, F.C.; Ostergaard, P.; Carver, C.; Brice, G.; Williams, N.; Mansour, S.; Mortimer, P.S.; Jeffery, S. Lymphoedema Consortium Analysis of the coding regions of VEGFR3 and VEGFC in Milroy disease and other primary lymphoedemas. Hum. Genet. 2009, 124, 625–631. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | NRP1 | NRP2 |
|---|---|---|
| Localization | 10p11.22 | 2q33.3 |
| Gene function | Protein coding | Protein coding |
| Expression in lymphatic system | Predominantly lymphatic valves | Small lymphatic vessels and capillaries |
| Lethality in mouse model | Embryo death at E8.5 (Nrp1−/−/Nrp2−/−) Embryo death at E10-E10.5 (Nrp1+/−/Nrp2−/−; Nrp1−/−/Nrp2+/−) | |
| Lymphatic phenotype in mouse model | Nrp1−/− malformation of valve precursor LECs [18] | Nrp2−/− reduction or loss of small lymphatic vessels and capillaries [19] Nrp2+/− and Vegfr3+/− abnormal lymphatic development and reduced lymphatic vessel branching [13] |
| NRP1 | MERGLPLLCAVLALVLAPAGAFRNDKCGDTIKIESPGYLTSPGYPHSYHPSEKCEWLIQAPDPYQRIMINFNPHFDLEDRDCKYDYVEVFDGENENGHFRGKFCGKIAPPPVVSSGPFLFIKFVSDYETHGAGFSIRYEIFKRGPECSQNYTTPSGVIKSPGFPEKYPNSLECTYIVFVPKMSEIILEFESFDLEPDSNPPGGMFCRYDRLEIWDGFPDVGPHIGRYCGQKTPGRIRSSSGILSMVFYTDSAIAKEGFSANYSVLQSSVSEDFKCMEALGMESGEIHSDQITASSQYSTNWSAERSRLNYPENGWTPGEDSYREWIQVDLGLLRFVTAVGTQGAISKETKKKYYVKTYKIDVSSNGEDWITIKEGNKPVLFQGNTNPTDVVVAVFPKPLITRFVRIKPATWETGISMRFEVYGCKITDYPCSGMLGMVSGLISDSQITSSNQGDRNWMPENIRLVTSRSGWALPPAPHSYINEWLQIDLGEEKIVRGIIIQGGKHRENKVFMRKFKIGYSNNGSDWKMIMDDSKRKAKSFEGNNNYDTPELRTFPALSTRFIRIYPERATHGGLGLRMELLGCEVEAPTAGPTTPNGNLVDECDDDQANCHSGTGDDFQLTGGTTVLATEKPTVIDSTIQSEFPTYGFNCEFGWGSHKTFCHWEHDNHVQLKWSVLTSKTGPIQDHTGDGNFIYSQADENQKGKVARLVSPVVYSQNSAHCMTFWYHMSGSHVGTLRVKLRYQKPEEYDQLVWMAIGHQGDHWKEGRVLLHKSLKLYQVIFEGEIGKGNLGGIAVDDISINNHISQEDCAKPADLDKKNPEIKIDETGSTPGYEGEGEGDKNISRKPGNVLKTLDPILITIIAMSALGVLLGAVCGVVLYCACWHNGMSERNLSALENYNFELVDGVKLKKDKLNTQSTYSEA |
| NRP2 | MDMFPLTWVFLALYFSRHQVRGQPDPPCGGRLNSKDAGYITSPGYPQDYPSHQNCEWIVYAPEPNQKIVLNFNPHFEIEKHDCKYDFIEIRDGDSESADLLGKHCGNIAPPTIISSGSMLYIKFTSDYARQGAGFSLRYEIFKTGSEDCSKNFTSPNGTIESPGFPEKYPHNLDCTFTILAKPKMEIILQFLIFDLEHDPLQVGEGDCKYDWLDIWDGIPHVGPLIGKYCGTKTPSELRSSTGILSLTFHTDMAVAKDGFSARYYLVHQEPLENFQCNVPLGMESGRIANEQISASSTYSDGRWTPQQSRLHGDDNGWTPNLDSNKEYLQVDLRFLTMLTAIATQGAISRETQNGYYVKSYKLEVSTNGEDWMVYRHGKNHKVFQANNDATEVVLNKLHAPLLTRFVRIRPQTWHSGIALRLELFGCRVTDAPCSNMLGMLSGLIADSQISASSTQEYLWSPSAARLVSSRSGWFPRIPQAQPGEEWLQVDLGTPKTVKGVIIQGARGGDSITAVEARAFVRKFKVSYSLNGKDWEYIQDPRTQQPKLFEGNMHYDTPDIRRFDPIPAQYVRVYPERWSPAGIGMRLEVLGCDWTDSKPTVETLGPTVKSEETTTPYPTEEEATECGENCSFEDDKDLQLPSGFNCNFDFLEEPCGWMYDHAKWLRTTWASSSSPNDRTFPDDRNFLRLQSDSQREGQYARLISPPVHLPRSPVCMEFQYQATGGRGVALQVVREASQESKLLWVIREDQGGEWKHGRIILPSYDMEYQIVFEGVIGKGRSGEIAIDDIRISTDVPLENCMEPISAFAGENFKVDIPEIHEREGYEDEIDDEYEVDWSNSSSATSGSGAPSTDKEKSWLYTLDPILITIIAMSSLGVLLGATCAGLLLYCTCSYSGLSSRSCTTLENYNFELYDGLKHKVKMNHQKCCSEA |
| Gene | Family | Pedigree | Sex | Age | Clinical Features | Age of Onset | Familial Case | Variant Nomenclature (NRP1: NM_003873.6; NP_001019799.1; NRP2: NM_003872.2; NP_003863.2) | dbSNP id | ACMG Classification | Frequency in GnomAD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NRP1 | 1 | Proband | M | 68 | lymphedema of the lower limbs | Juvenile | NO | c.2557A > C; p.Ile853Leu | rs760388137 | Likely benign | 0.0001178 |
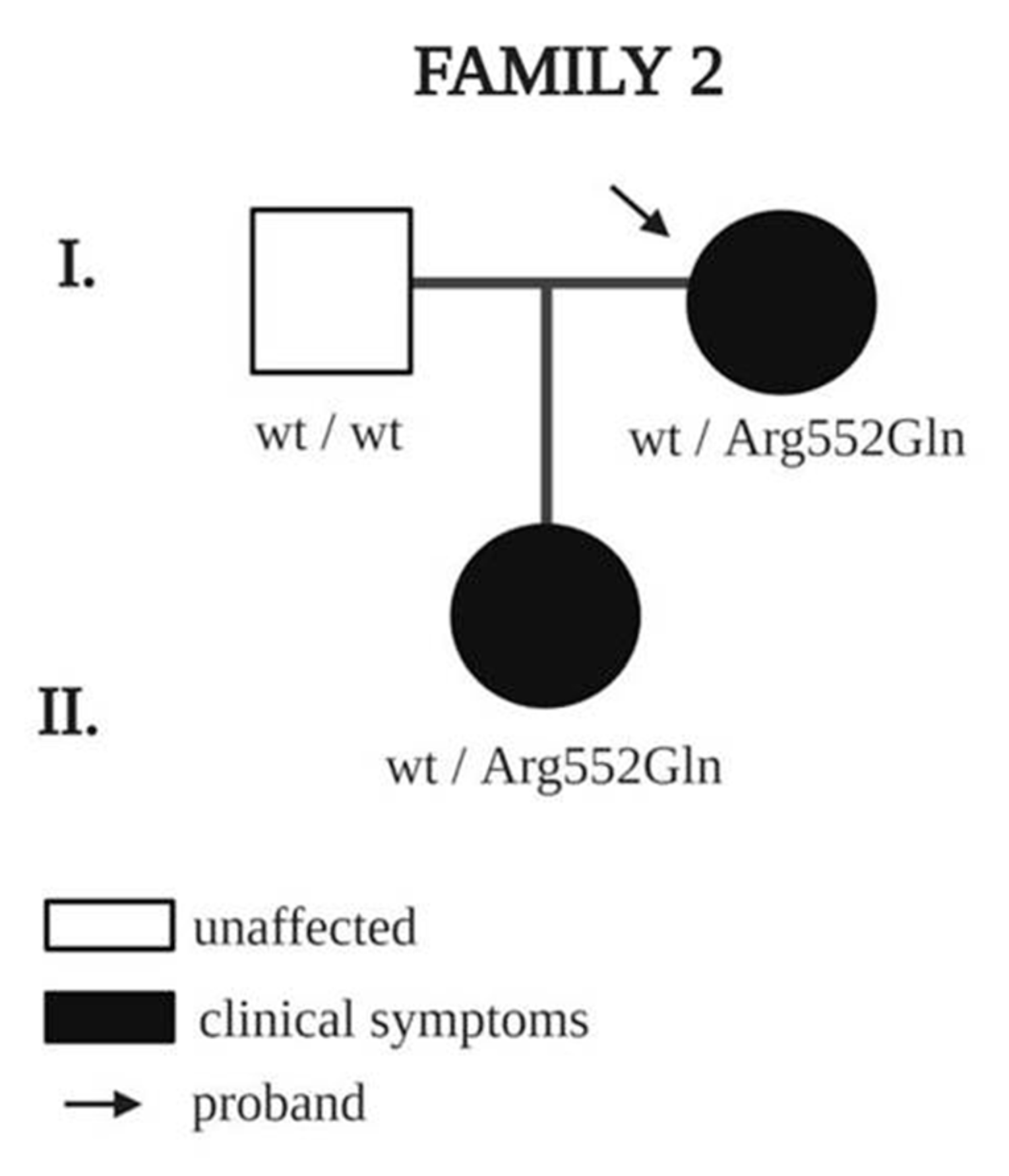
| NRP1 | 2 | Proband | F | 54 | edema of left foot and ankle | 27 | YES | c.1655G > A; p.Arg552Gln | rs757990959 | VUS | 0.00004873 |
| NRP1 | 2 | Daughter | F | 21 | lymphedema of left lower limb | 10 | YES | c.1655G > A; p.Arg552Gln | |||
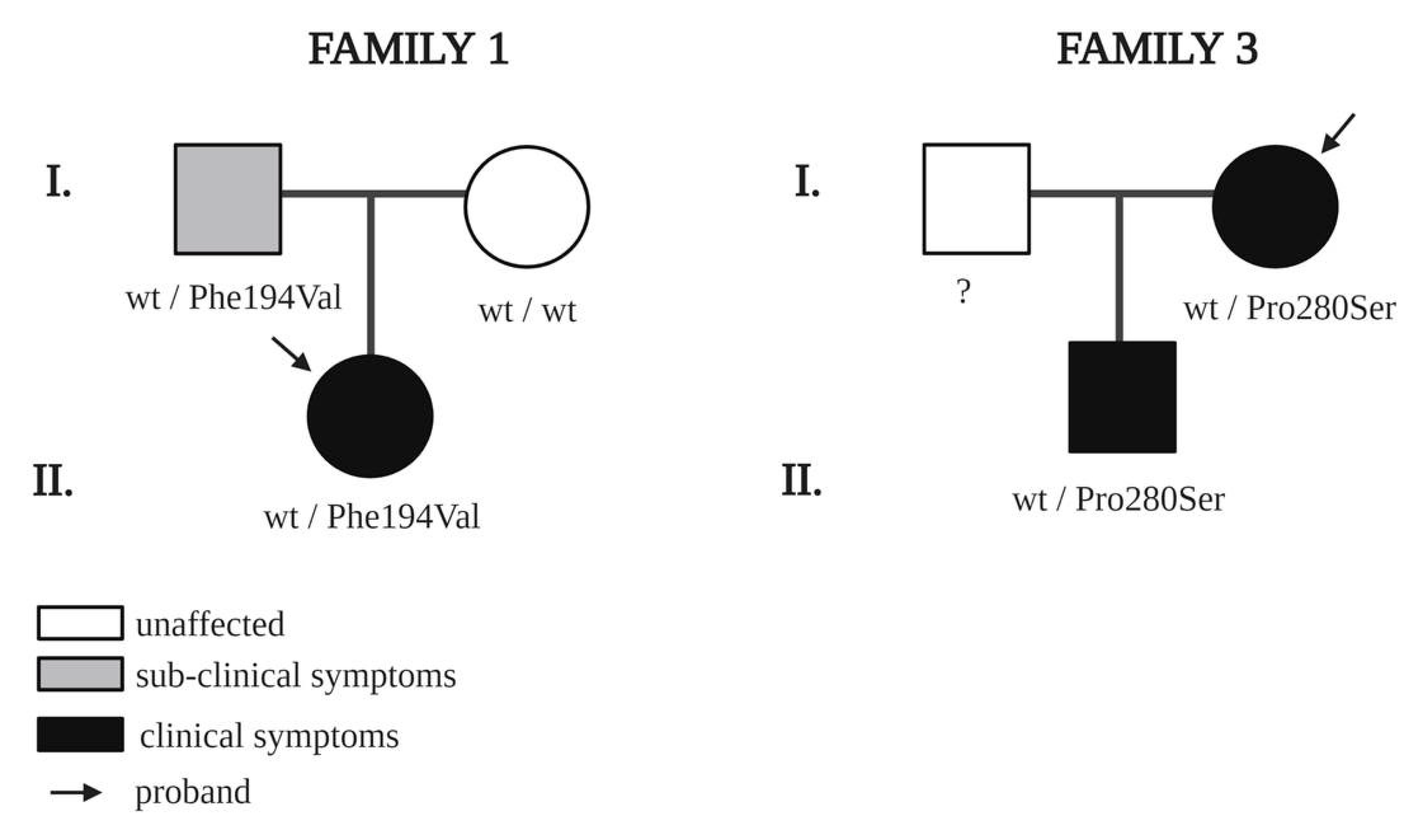
| NRP2 | 1 | Proband | F | 18 | edema of left hand | Congenital | NO | c.580T > G; p.Phe194Val | rs755679361 | VUS | 0.000008122 |
| NRP2 | 1 | Mother | F | 44 | Healthy | / | NO | - | |||
| NRP2 | 1 | Father | M | 46 | Apparently healthy but with subclinical symptoms | / | NO | c.580T > G; p.Phe194Val | |||
| NRP2 | 2 | Proband | M | 9 | edema of left foot, ankle and heel since vaccination | 3 months | NO | c.1748T > C; p.Ile583Thr | rs746130411 | VUS | 0.000004068 |
| NRP2 | 3 | Proband | F | 45 | edema of right foot | 32 | YES | c.838C > T; p.Pro280Ser | rs79750907 | Likely benign | 0.001783 |
| NRP2 | 3 | Son | M | 15 | edema of left foot | 9 | YES | c.838C > T; p.Pro280Ser | |||
| NRP2 | 4 | Proband | F | 50 | swelling of lower limbs | 42 | NO | c.1000C > T; p.Arg334Cys | rs114144673 | Likely benign | 0.001525 |
| Template | Seq Identity | Oligo State | QSQE | Found by | Method | Resolution | Seq Similarity | Coverage | Description | |
|---|---|---|---|---|---|---|---|---|---|---|
| NRP1 | 4gz9.1.A | 91.50 | monomer | - | BLAST | X-ray | 2.70Å | 0.60 | 0.61 | Neuropilin-1 |
| 4gz9.1.A | 91.50 | monomer | - | HHBlits | X-ray | 2.70Å | 0.60 | 0.61 | Neuropilin-1 | |
| 2qql.1.A | 51.95 | Homodimer | 0.37 | HHBlits | X-ray | 3.10Å | 0.46 | 0.61 | Neuropilin-2 | |
| 2qql.1.A | 53.14 | Homodimer | 0.35 | BLAST | X-ray | 3.10Å | 0.47 | 0.60 | Neuropilin-2 | |
| 2qqk.1.A | 51.95 | Monomer | - | HHBlits | X-ray | 2.75Å | 0.48 | 0.61 | Neuropilin-2 | |
| 2qqk.1.A | 53.14 | Monomer | - | BLAST | X-ray | 2.75Å | 0.47 | 0.60 | Neuropilin-2 | |
| 2qqm.1.A | 99.78 | monomer | - | BLAST | X-ray | 2.00Å | 0.62 | 0.48 | Neuropilin-1 | |
| 2qqm.1.A | 99.78 | monomer | - | HHBlits | X-ray | 2.00Å | 0.62 | 0.48 | Neuropilin-1 | |
| 2qqo.1.A | 51.36 | monomer | - | HHBlits | X-ray | 2.30Å | 0.46 | 0.48 | Neuropilin-2 | |
| 2qqo.1.A | 52.51 | monomer | - | BLAST | X-ray | 2.30Å | 0.46 | 0.47 | Neuropilin-2 | |
| NRP2 | 2qql.1.A | 100.00 | Homo-monomer | 0.51 | BLAST | X-ray | 3.10 Å | 0.62 | 0.62 | Neuropilin-2 |
| 2qqk.1.A | 100.00 | monomer | - | BLAST | X-ray | 2.75Å | 0.62 | 0.62 | Neuropilin-2 | |
| 2qql.1.A | 100.00 | Homodimer | 0.51 | HHBlits | X-ray | 3.10Å | 0.62 | 0.61 | Neuropilin-2 | |
| 2qqk.1.A | 100.00 | monomer | - | HHBlits | X-ray | 2.75Å | 0.62 | 0.61 | Neuropilin-2 | |
| 2qqo.1.A | 100.00 | Monomer | - | HHBlits | X-ray | 2.30Å | 0.62 | 0.48 | Neuropilin-2 | |
| 2qqm.1.A | 51.81 | Monomer | - | HHBlits | X-ray | 2.00Å | 0.46 | 0.47 | Neuropilin-1 | |
| 2qqm.1.A | 53.05 | monomer | - | BLAST | X-ray | 2.00Å | 0.46 | 0.48 | Neuropilin-1 | |
| 2qqj.1.A | 100.00 | monomer | - | BLAST | X-ray | 1.95Å | 0.62 | 0.34 | Neuropilin-2 | |
| 2qqj.1.A | 100.00 | monomer | - | HHBlits | X-ray | 1.95Å | 0.62 | 0.34 | Neuropilin-2 | |
| 2qqi.1.A | 51.77 | monomer | - | BLAST | X-ray | 1.80Å | 0.46 | 0.33 | Neuropilin-1 |
| Variant | Amino Acid | Molecular Interactions | Bond Length in Angstroms | Bond Type | |
|---|---|---|---|---|---|
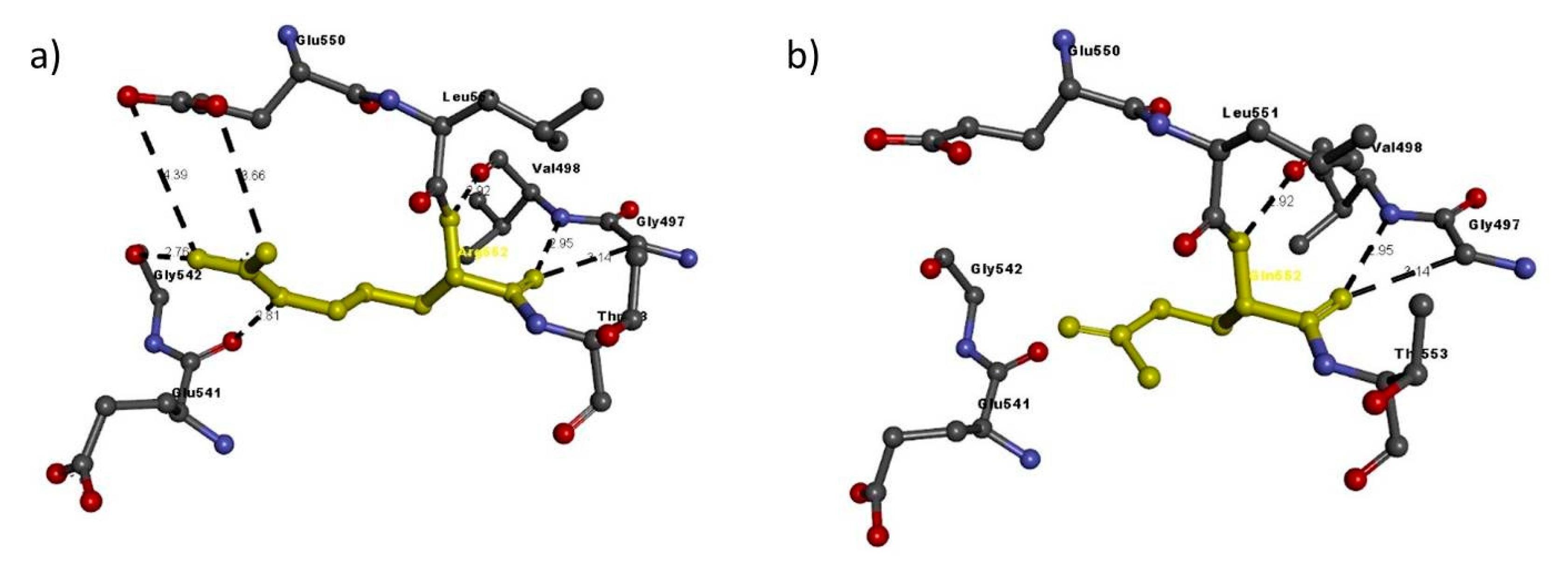
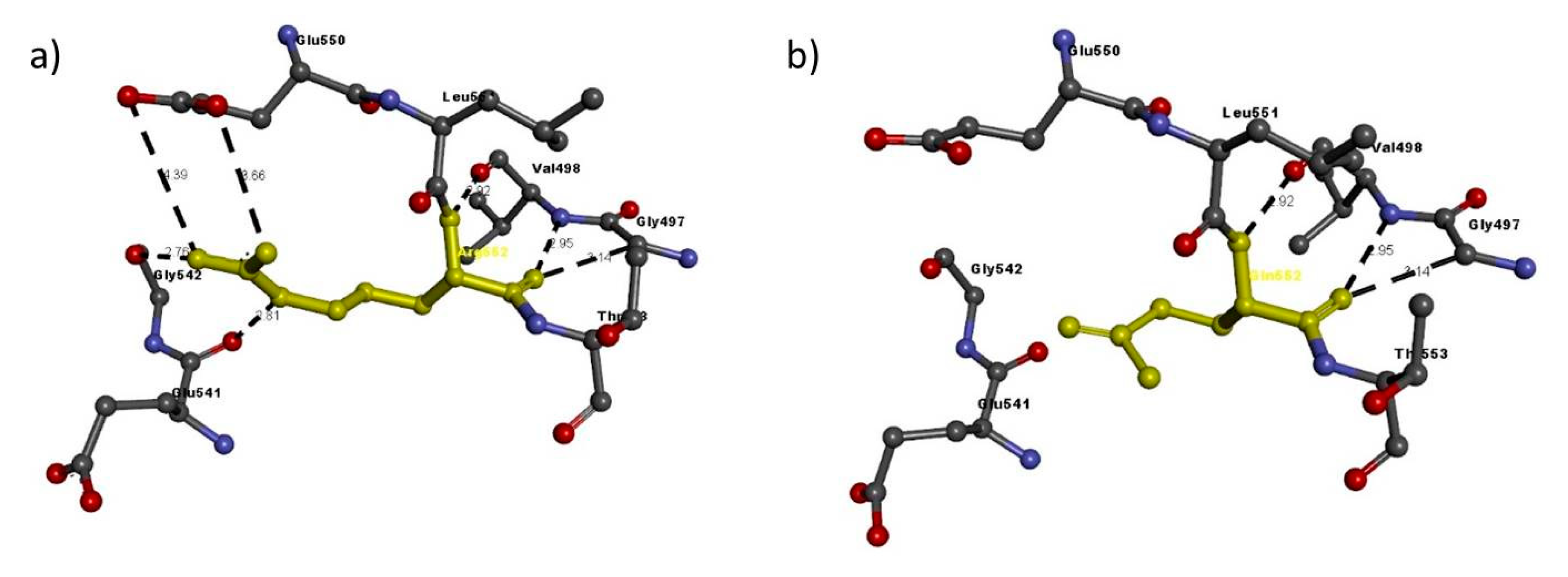
| NRP1 FAM2 | Arg552Gln | Arg552 | Arg552: N–Glu550: O | 3.66 | H-bond |
| Arg552: N–Glu550: O | 4.39 | H-bond | |||
| Val498: N–Arg552: O | 2.95 | H-bond | |||
| Arg552: N–Val498: O | 2.92 | H-bond | |||
| Arg552: N–Glu541: O | 2.81 | H-bond | |||
| Arg552: N–Gly542: O | 2.76 | H-bond | |||
| Gly497: C–Arg552: O | 3.14 | H-bond | |||
| Gln552 | Val498: N–Gln552: O | 2.95 | H-bond | ||
| Gln552: N–Val498: O | 2.92 | H-bond | |||
| Gly497: C–Gln552: O | 3.14 | H-bond |
| Variant | Amino Acid | Molecular Interactions | Bond Length in Angstroms | Bond Type | |
|---|---|---|---|---|---|
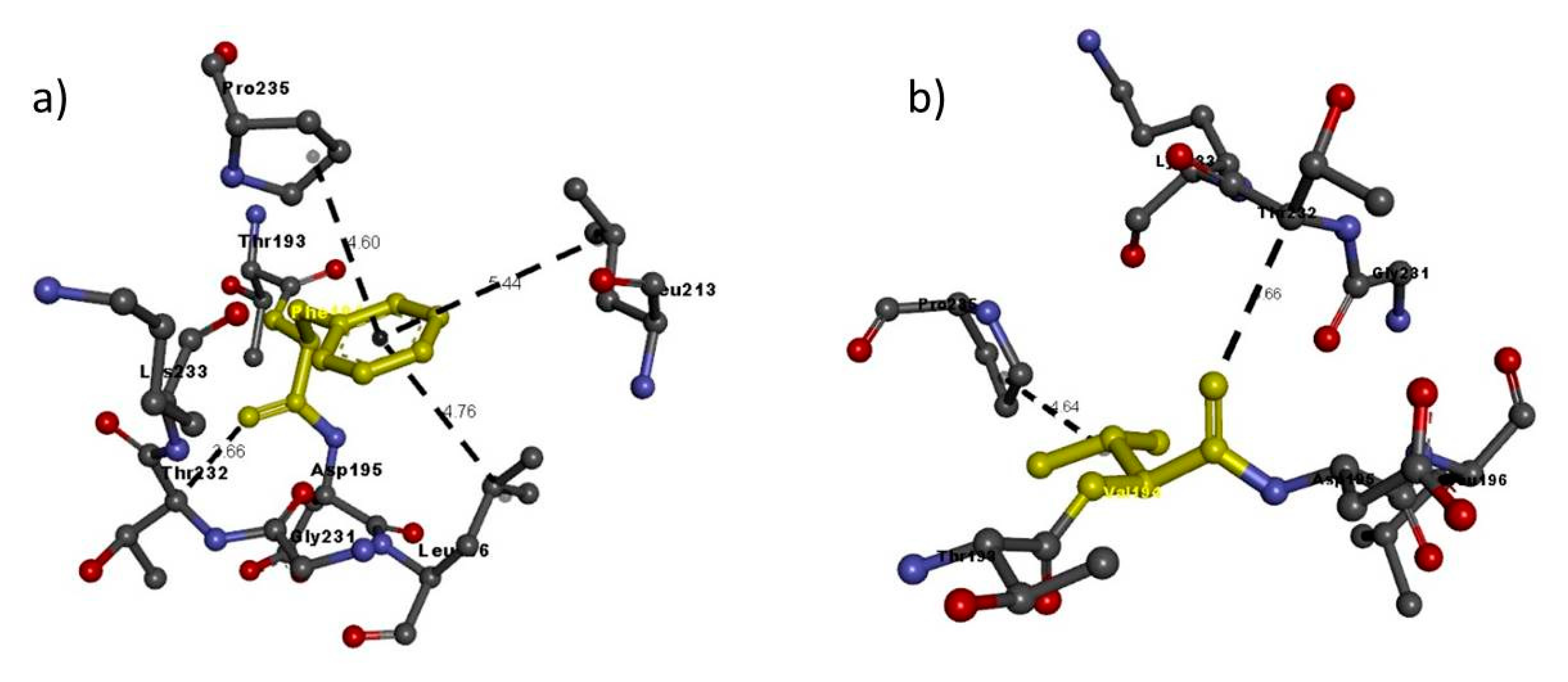
| NRP2 FAM 1 | Phe194Val | Phe194 | Thr232: C–Phe194: O | 3.66 | H-bond |
| Phe194: A–Phe196 | 4.76 | H-bond | |||
| Phe1941: A–Phe213 | 5.44 | Pi interaction | |||
| Phe194: A–Pro235 | 4.60 | Pi interaction | |||
| Val194 | Thr232: C–Val194: O | 3.66 | H-bond | ||
| Val194: A–Pro235 | 4.64 | Hydrophobic interaction | |||
| NRP2 FAM 3 | Pro280Ser | Pro280 | Phe425–Pro280 | 4.99 | Pi interaction |
| Ser280 | No bonds | - | - | ||
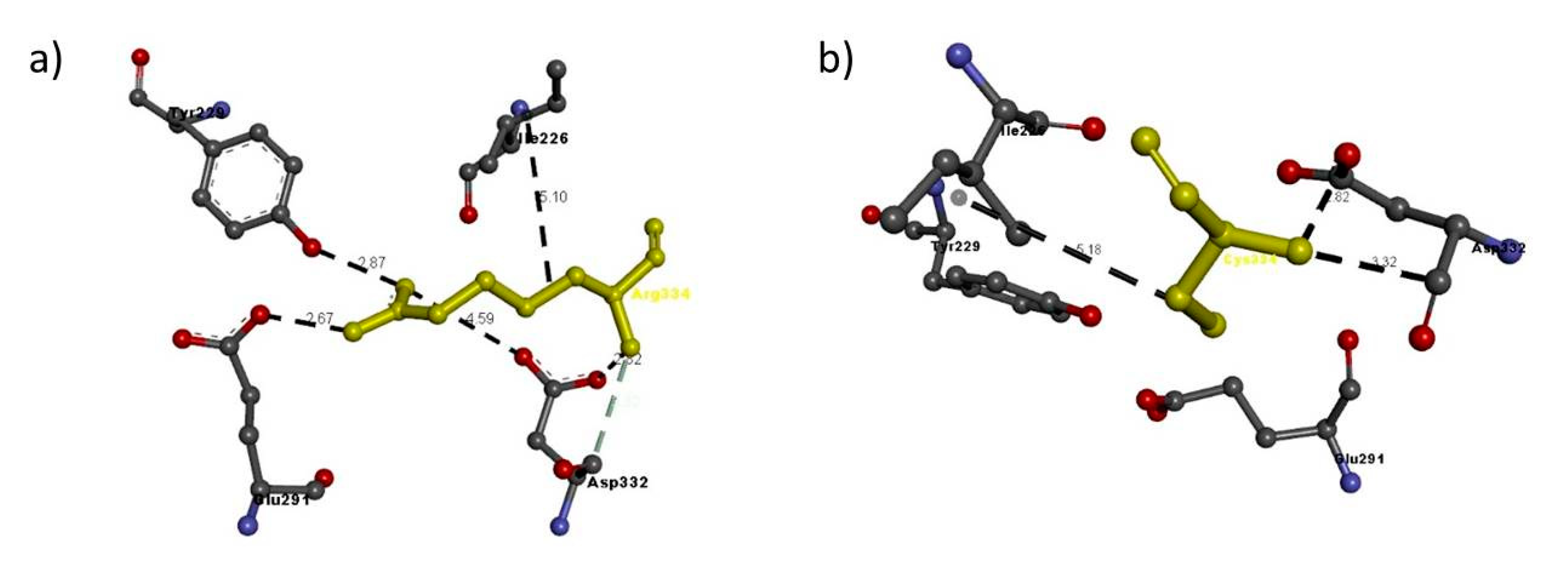
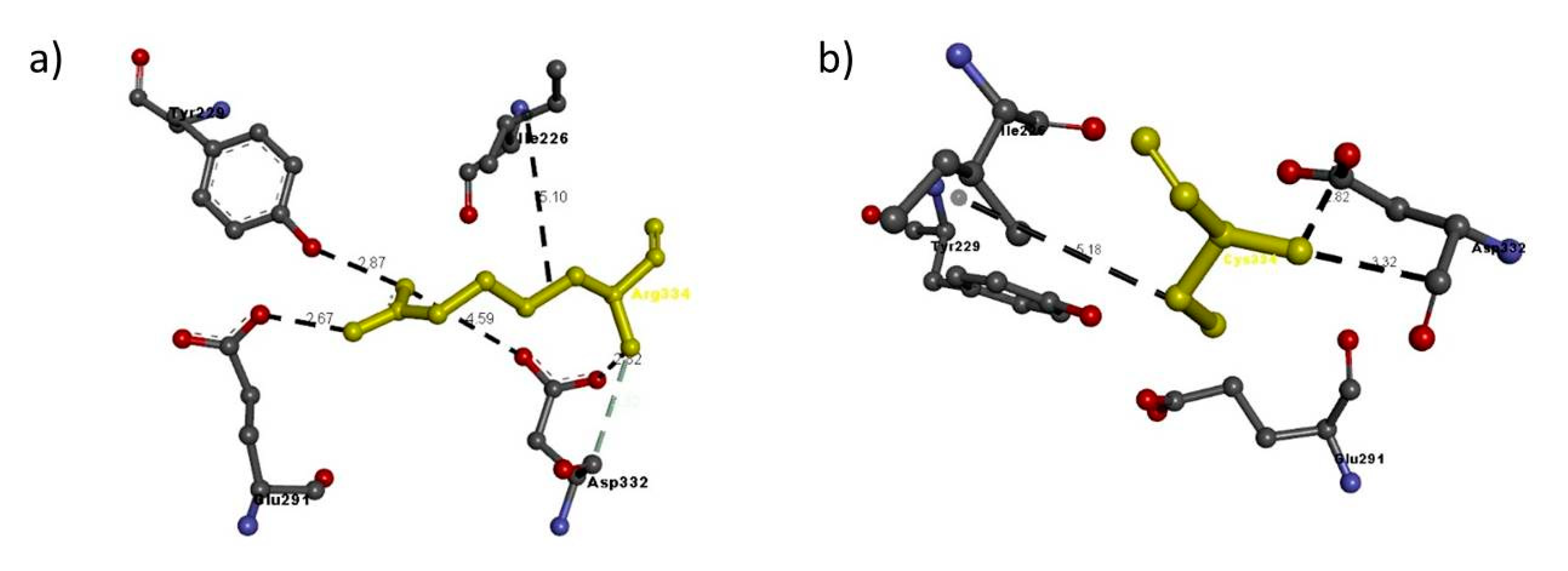
| NRP2 FAM 4 | Arg334Cys | Arg334 | Arg334: N–Glu291: O | 2.67 | H-bond |
| Arg334: N–Asp332: O | 4.59 | H-bond | |||
| Arg334: N–Asp332: O | 2.82 | H-bond | |||
| Arg334: N–Tyr229: O | 2.87 | H-bond | |||
| Asp332: C–Arg334: N | 3.32 | H-bond | |||
| Arg334–Ile226 | 5.10 | Hydrophobic interaction | |||
| Cys334 | Cys334: N - Asp332: O | 2.82 | H-bond | ||
| Asp332: C–A:Cys334:N | 3.32 | H-bond | |||
| A:Cys334–A:Ile226 | 5.18 | Hydrophobic interaction | |||
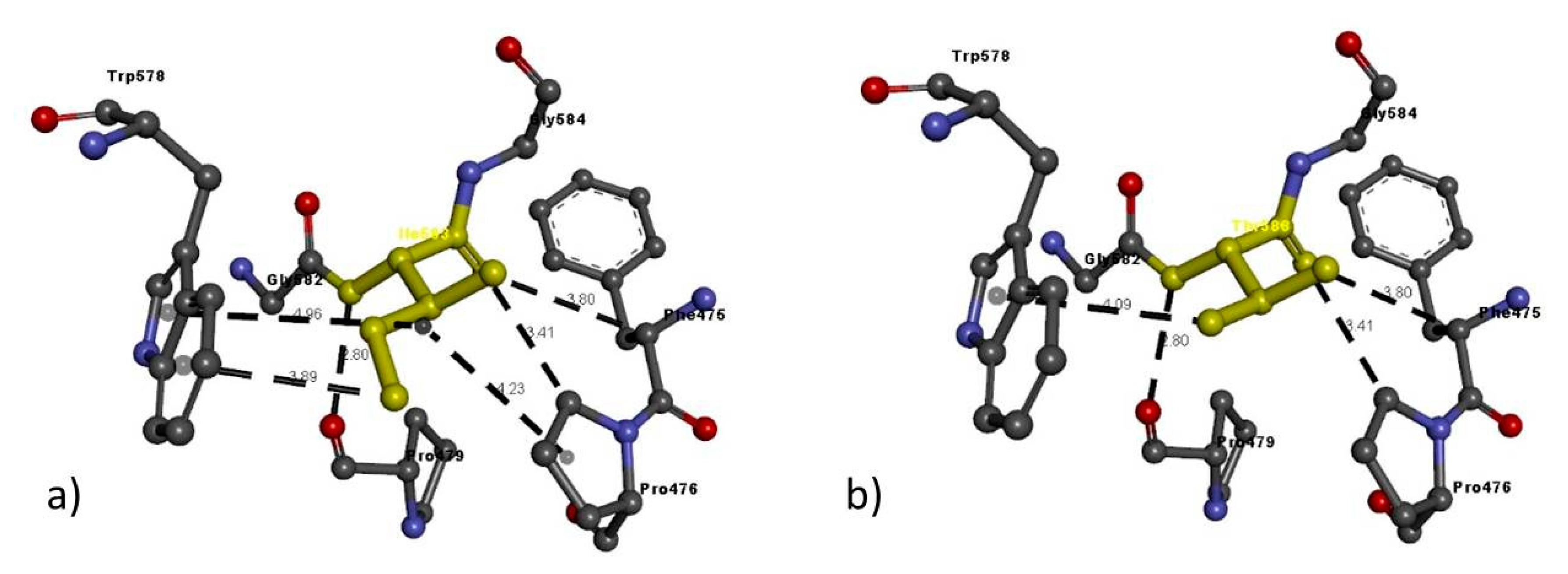
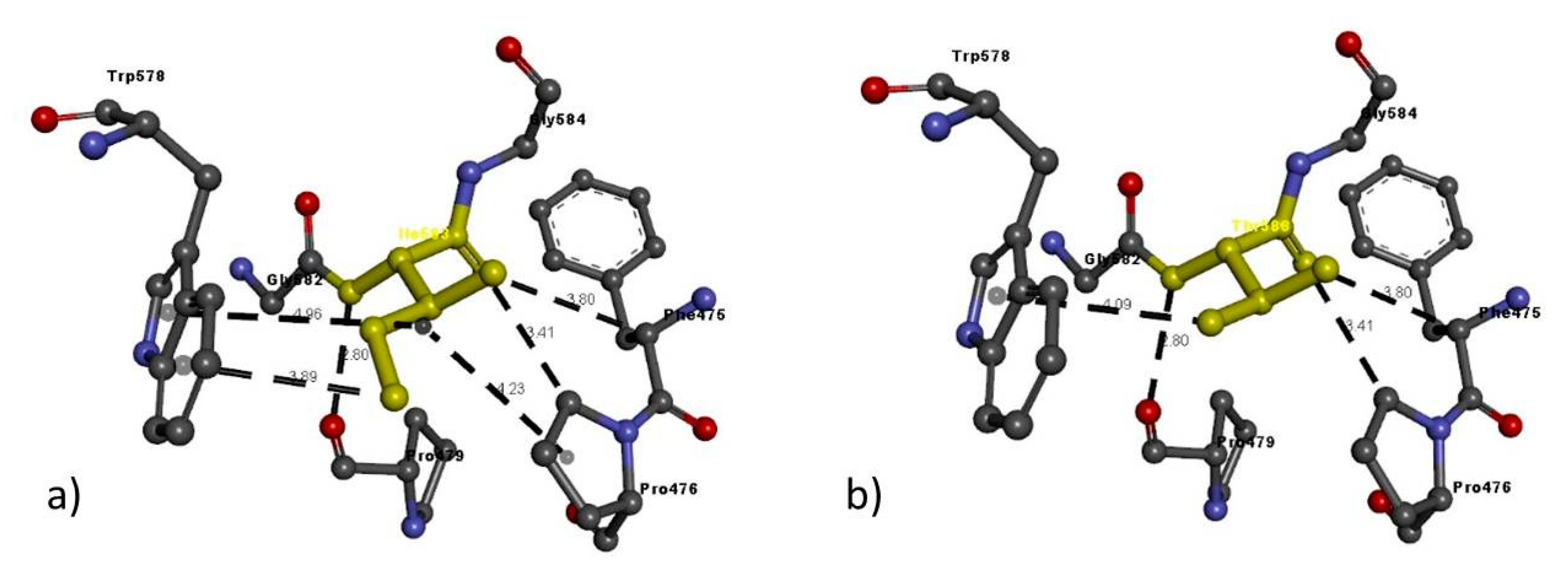
| NRP2 FAM 2 | Ile583Thr | Ile583 | Ile583: N–Pro479: O | 2.80 | H-bond |
| Phe475: C–Ile583: O | 3.80 | H-bond | |||
| Pro476: C–Ile583: O | 3.41 | H-bond | |||
| Ile583: C–Trp578 | 3.89 | Pi interactions | |||
| Pro476: A–Ile583 | 4.23 | Pi interactions | |||
| Trp578: A–Ile583 | 4.91 | Pi interactions | |||
| Thr583 | Thr583: N–Pro479: O | 2.80 | H-bond | ||
| Phe475: C–Thr583: O | 3.80 | H-bond | |||
| Pro476: C–Thr583: O | 3.41 | H-bond | |||
| Thr583: O–Trp578 | 4.09 | Pi interactions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelini, S.; Amato, B.; Ricci, M.; Kenanoglu, S.; Veselenyiova, D.; Kurti, D.; Baglivo, M.; Manara, E.; Dundar, M.; Krajcovic, J.; et al. Segregation Analysis of Rare NRP1 and NRP2 Variants in Families with Lymphedema. Genes 2020, 11, 1361. https://doi.org/10.3390/genes11111361
Michelini S, Amato B, Ricci M, Kenanoglu S, Veselenyiova D, Kurti D, Baglivo M, Manara E, Dundar M, Krajcovic J, et al. Segregation Analysis of Rare NRP1 and NRP2 Variants in Families with Lymphedema. Genes. 2020; 11(11):1361. https://doi.org/10.3390/genes11111361
Chicago/Turabian StyleMichelini, Sandro, Bruno Amato, Maurizio Ricci, Sercan Kenanoglu, Dominika Veselenyiova, Danjela Kurti, Mirko Baglivo, Elena Manara, Munis Dundar, Juraj Krajcovic, and et al. 2020. "Segregation Analysis of Rare NRP1 and NRP2 Variants in Families with Lymphedema" Genes 11, no. 11: 1361. https://doi.org/10.3390/genes11111361
APA StyleMichelini, S., Amato, B., Ricci, M., Kenanoglu, S., Veselenyiova, D., Kurti, D., Baglivo, M., Manara, E., Dundar, M., Krajcovic, J., Basha, S. H., Priya, S., Serrani, R., Miggiano, G. A. D., Aquilanti, B., Matera, G., Velluti, V., Gagliardi, L., Dautaj, A., & Bertelli, M. (2020). Segregation Analysis of Rare NRP1 and NRP2 Variants in Families with Lymphedema. Genes, 11(11), 1361. https://doi.org/10.3390/genes11111361