Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment

 , , ,
, , ,  ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Participants’ Recruitment
2.3. Whole Exome Sequencing and Data Analysis
2.4. Annotation and Filtering Strategy
2.5. Sanger Sequencing
2.6. Evolutionary Conservation of Amino Acids and Secondary Structure Analysis
2.7. Protein Modelling
3. Results
3.1. Participants Phenotypes
3.2. WES Identification of Candidate Gene and Variants
3.3. Sanger Sequencing Confirmation of Variants
3.4. Analysis of the CLIC5—NM_016929.5(CLIC5):p.(L75P) Variant on the Protein
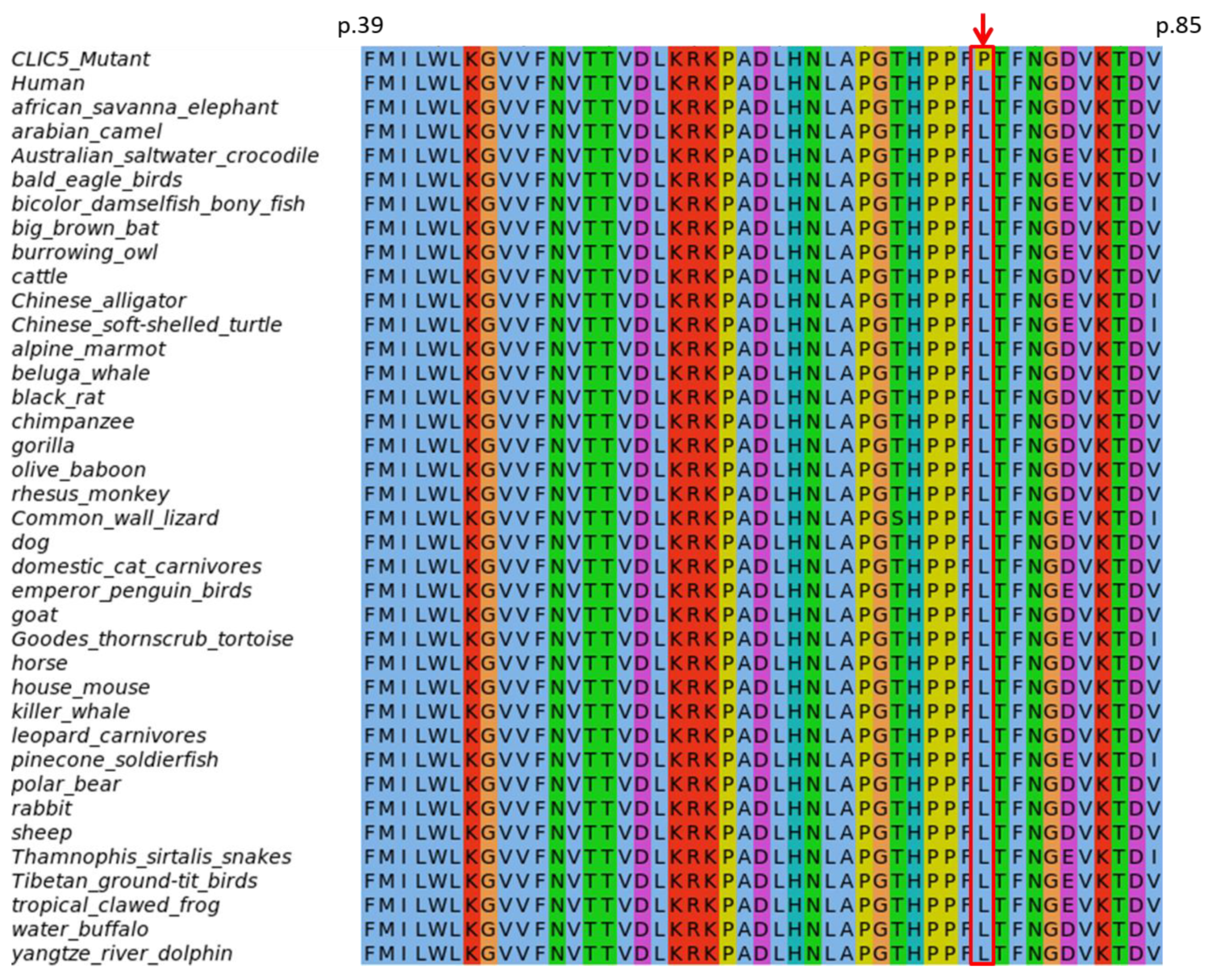
3.4.1. Evolutionary Conservation of Amino Acids
3.4.2. Protein Modelling: Secondary Structure Analysis and Domain Search
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Web Resources
References
- Olusanya, B.O.; Neumann, K.J.; Saunders, J.E. The global burden of disabling hearing impairment: A call to action. Bull. World Health Organ. 2014, 92, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, I.; Liaqat, K.; Schatteman, I.; Bharadwaj, T.; Nasir, A.; Acharya, A.; Ahmad, W.; Van Camp, G.; Leal, S.M. Autosomal dominantly inherited GREB1L variants in individuals with profound sensorineural hearing impairment. Genes 2020, 11, 687. [Google Scholar] [CrossRef] [PubMed]
- Wonkam Tingang, E.; Noubiap, J.J.; Fokouo, J.V.F.; Oluwole, O.G.; Nguefack, S.; Chimusa, E.R.; Wonkam, A. Hearing impairment overview in Africa: The case of Cameroon. Genes 2020, 11, 233. [Google Scholar] [CrossRef] [PubMed]
- Lebeko, K.; Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Wonkam, A. Genetics of hearing loss in Africans: Use of next generation sequencing is the best way forward. Pan Afr. Med. J. 2015, 20. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary hearing loss and deafness overview. In GeneReviews®[Internet]; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Tingang Wonkam, E.; Chimusa, E.; Noubiap, J.J.; Adadey, S.M.; Fokouo, J.V.F.; Wonkam, A. GJB2 and GJB6 mutations in hereditary recessive non-syndromic hearing impairment in Cameroon. Genes 2019, 10, 844. [Google Scholar] [CrossRef]
- Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Makubalo, N.; Wright, G.; Entfellner, J.-B.D.; Tiffin, N.; Wonkam, A. Sequencing of GJB2 in Cameroonians and Black South Africans and comparison to 1000 Genomes project data support need to revise strategy for discovery of nonsyndromic deafness genes in Africans. OMICS 2014, 18, 705–710. [Google Scholar] [CrossRef]
- Bosch, J.; Lebeko, K.; Nziale, J.J.N.; Dandara, C.; Makubalo, N.; Wonkam, A. In search of genetic markers for nonsyndromic deafness in Africa: A study in Cameroonians and Black South Africans with the GJB6 and GJA1 Candidate Genes. OMICS 2014, 18, 481–485. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Yan, D.; Tekin, D.; Bademci, G.; Foster, J.; Cengiz, F.B.; Kannan-Sundhari, A.; Guo, S.; Mittal, R.; Zou, B.; Grati, M.; et al. Spectrum of DNA variants for nonsyndromic deafness in a large cohort from multiple continents. Hum. Genet. 2016, 135, 953–961. [Google Scholar] [CrossRef]
- Lebeko, K.; Sloan-Heggen, C.M.; Noubiap, J.J.N.; Dandara, C.; Kolbe, D.L.; Ephraim, S.S.; Booth, K.T.; Azaiez, H.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet. 2016, 90, 288–290. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Chakchouk, I.; Zhang, D.; Zhang, Z.; Francioli, L.C.; Santos-Cortez, R.L.P.; Schrauwen, I.; Leal, S.M. Disparities in discovery of pathogenic variants for autosomal recessive non-syndromic hearing impairment by ancestry. Eur. J. Hum. Genet. 2019, 27, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Seco, C.Z.; Oonk, A.M.; Domínguez-Ruiz, M.; Draaisma, J.M.; Gandía, M.; Oostrik, J.; Neveling, K.; Kunst, H.P.; Hoefsloot, L.H.; del Castillo, I.; et al. Progressive hearing loss and vestibular dysfunction caused by a homozygous nonsense mutation in CLIC5. Eur. J. Hum. Genet. 2015, 23, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, L.H.; Longo-Guess, C.M.; Berryman, M.; Shin, J.-B.; Saylor, K.W.; Yu, H.; Gillespie, P.G.; Johnson, K.R. The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J. Neurosci. 2006, 26, 10188–10198. [Google Scholar] [CrossRef]
- Wonkam, A.; Noubiap, J.J.N.; Djomou, F.; Fieggen, K.; Njock, R.; Toure, G.B. Aetiology of childhood hearing loss in Cameroon (sub-Saharan Africa). Eur. J. Med. Genet. 2013, 56, 20–25. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4. [Google Scholar] [CrossRef]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human non-synonymous and splice site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structuRes. and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Mi, W.; Liang, Y.-H.; Li, L.; Su, X.-D. The crystal structure of human chloride intracellular channel protein 2: A disulfide bond with functional implications. Proteins Struct. Funct. Bioinform. 2008, 71, 509–513. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]
- Pierchala, B.A.; Muñoz, M.R.; Tsui, C.C. Proteomic analysis of the slit diaphragm complex: CLIC5 is a protein critical for podocyte morphology and function. Kidney Int. 2010, 78, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Wegner, B.; Al-Momany, A.; Kulak, S.C.; Kozlowski, K.; Obeidat, M.; Jahroudi, N.; Paes, J.; Berryman, M.; Ballermann, B.J. CLIC5A, a component of the ezrin-podocalyxin complex in glomeruli, is a determinant of podocyte integrity. Am. J. Physiol. Ren. Physiol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Bradford, E.M.; Miller, M.L.; Prasad, V.; Nieman, M.L.; Gawenis, L.R.; Berryman, M.; Lorenz, J.N.; Tso, P.; Shull, G.E. CLIC5 mutant mice are resistant to diet-induced obesity and exhibit gastric hemorrhaging and increased susceptibility to torpor. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1531–R1542. [Google Scholar] [CrossRef][Green Version]
- Gururaja Rao, S.; Patel, N.J.; Singh, H. Intracellular chloride channels: Novel biomarkers in diseases. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Salles, F.T.; Andrade, L.R.; Tanda, S.; Grati, M.; Plona, K.L.; Gagnon, L.H.; Johnson, K.R.; Kachar, B.; Berryman, M.A. CLIC5 stabilizes membrane-actin filament linkages at the base of hair cell stereocilia in a molecular complex with radixin, taperin, and myosin VI. Cytoskeleton 2014, 71, 61–78. [Google Scholar] [CrossRef]
- Breuza, L.; Poux, S.; Estreicher, A.; Famiglietti, M.L.; Magrane, M.; Tognolli, M.; Bridge, A.; Baratin, D.; Redaschi, N. The UniProt Consortium. The UniProtKB guide to the human proteome. Database 2016, 2016, bav120. [Google Scholar] [CrossRef]
- Schrauwen, I.; Hasin-Brumshtein, Y.; Corneveaux, J.J.; Ohmen, J.; White, C.; Allen, A.N.; Lusis, A.J.; Van Camp, G.; Huentelman, M.J.; Friedman, R.A. A comprehensive catalogue of the coding and non-coding transcripts of the human inner ear. Hear. Res. 2016, 333, 266–274. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wonkam-Tingang, E.; Schrauwen, I.; Esoh, K.K.; Bharadwaj, T.; Nouel-Saied, L.M.; Acharya, A.; Nasir, A.; Adadey, S.M.; Mowla, S.; Leal, S.M.; et al. Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment. Genes 2020, 11, 1249. https://doi.org/10.3390/genes11111249
Wonkam-Tingang E, Schrauwen I, Esoh KK, Bharadwaj T, Nouel-Saied LM, Acharya A, Nasir A, Adadey SM, Mowla S, Leal SM, et al. Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment. Genes. 2020; 11(11):1249. https://doi.org/10.3390/genes11111249
Chicago/Turabian StyleWonkam-Tingang, Edmond, Isabelle Schrauwen, Kevin K. Esoh, Thashi Bharadwaj, Liz M. Nouel-Saied, Anushree Acharya, Abdul Nasir, Samuel M. Adadey, Shaheen Mowla, Suzanne M. Leal, and et al. 2020. "Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment" Genes 11, no. 11: 1249. https://doi.org/10.3390/genes11111249
APA StyleWonkam-Tingang, E., Schrauwen, I., Esoh, K. K., Bharadwaj, T., Nouel-Saied, L. M., Acharya, A., Nasir, A., Adadey, S. M., Mowla, S., Leal, S. M., & Wonkam, A. (2020). Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment. Genes, 11(11), 1249. https://doi.org/10.3390/genes11111249