Genetics and Genomics of Pediatric Pulmonary Arterial Hypertension


Abstract
1. Introduction
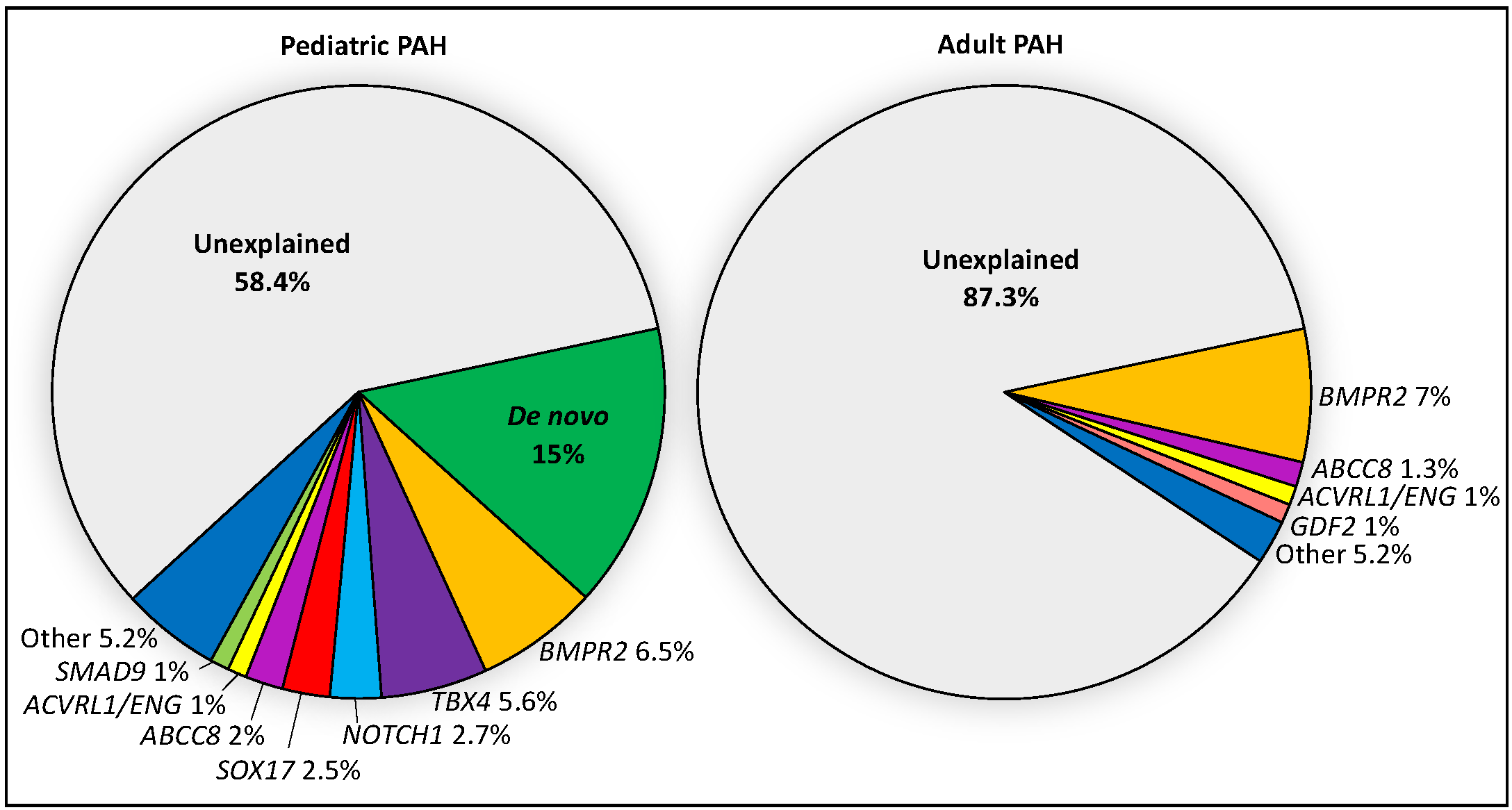
2. Genetics of Pediatric PAH—Current Knowledge
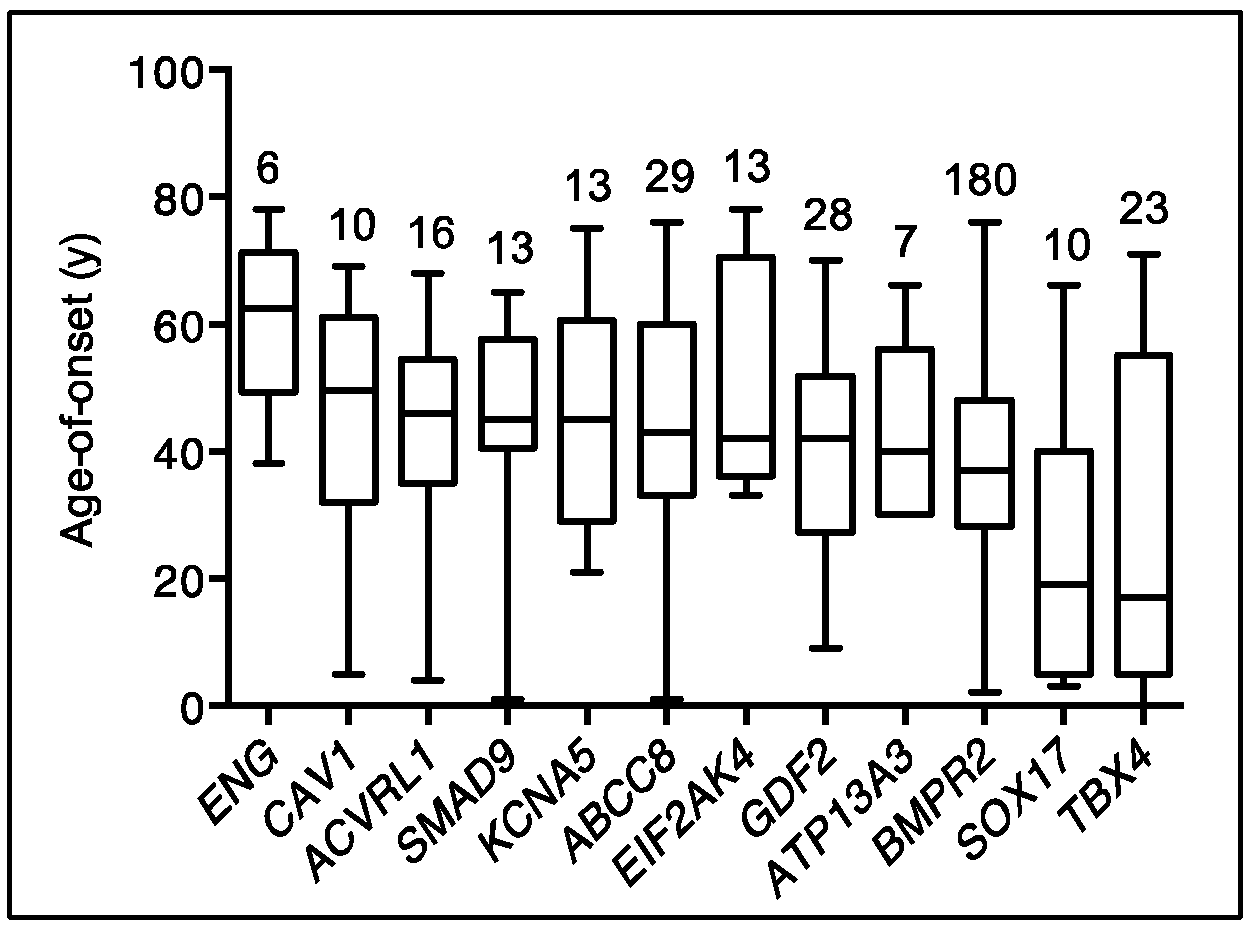
2.1. TBX4
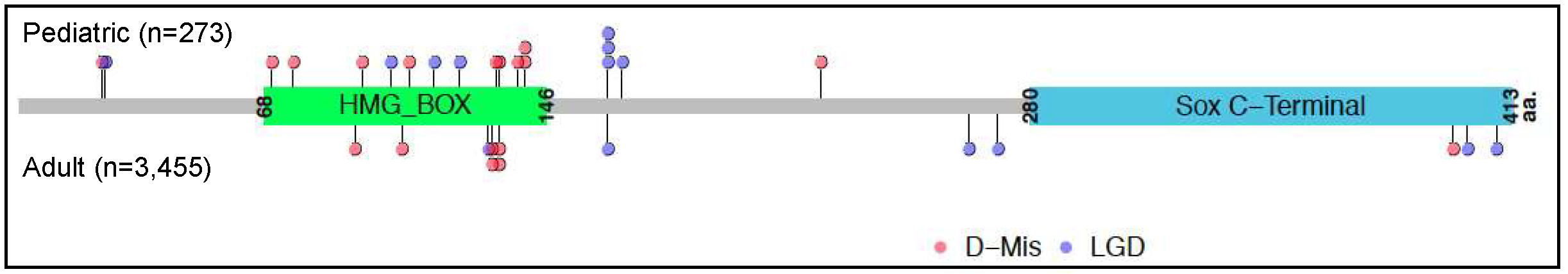
2.2. SOX17
2.3. The Relative Contribution of Other Known PAH Risk Genes
2.4. De Novo Variants
2.5. Genetic Ancestry
2.6. The Role of Other “Omics” in PAH
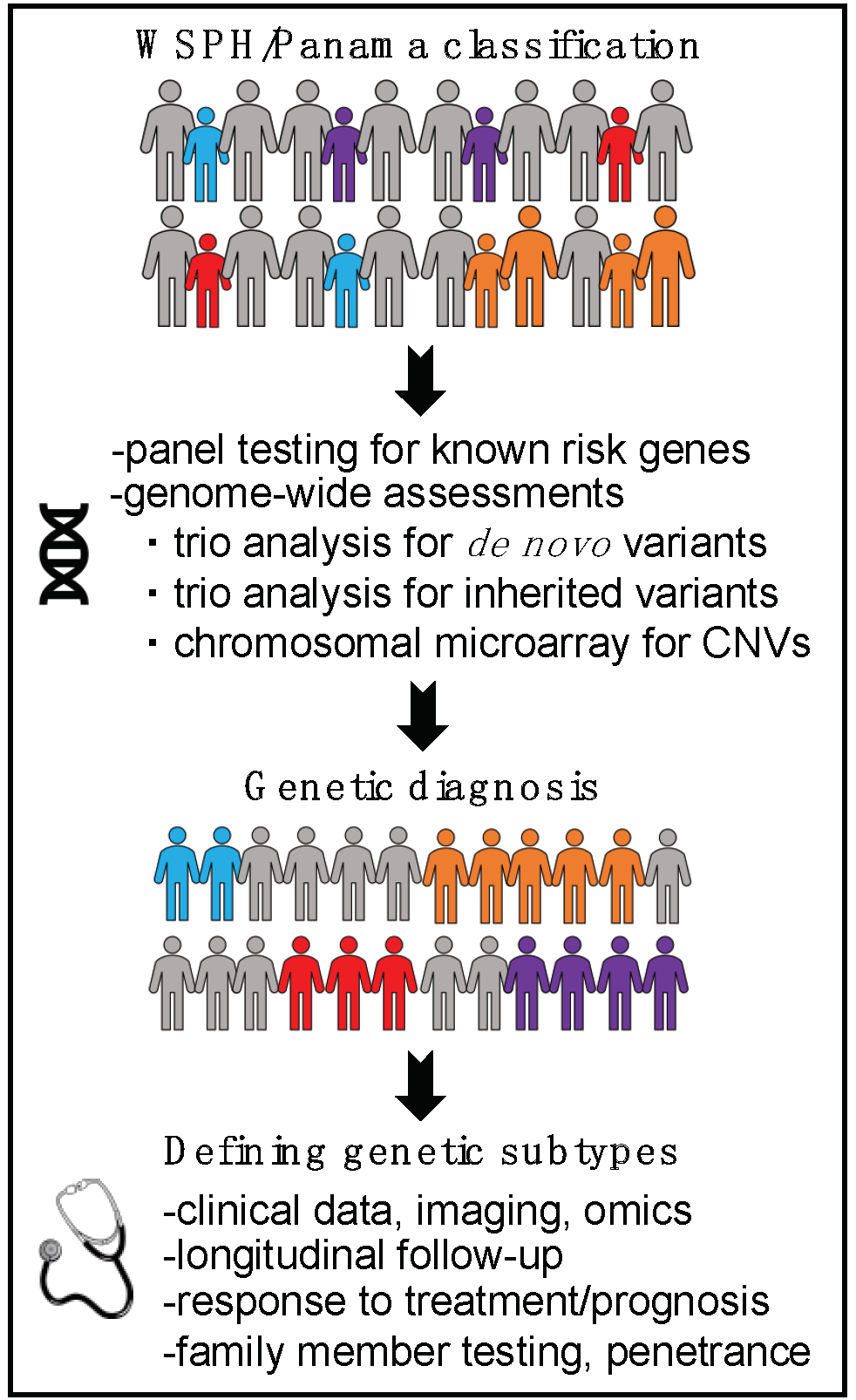
3. A Genomics First Approach towards Better Understanding of Pediatric PAH
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, L.; Jick, S.S.; Breitenstein, S.; Hernandez, G.; Michel, A.; Vizcaya, D. Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm. Circ. 2017, 7, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [PubMed]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Palomares, A.; Gu, M.; Grubert, F.; Berest, I.; Sa, S.; Kasowski, M.; Arnold, C.; Shuai, M.; Srivas, R.; Miao, S.; et al. Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension. Nat. Commun. 2020, 11, 1673. [Google Scholar] [CrossRef]
- Hansmann, G. Pulmonary hypertension in infants, children, and young adults. J. Am. Coll. Cardiol. 2017, 69, 2551–2569. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 2019, 53, 1801916. [Google Scholar] [CrossRef]
- Saji, T. Update on pediatric pulmonary arterial hypertension. Differences and similarities to adult disease. Circ. J. 2013, 77, 2639–2650. [Google Scholar] [CrossRef]
- Zhu, N.; Swietlik, E.M.; Welch, C.L.; Pauciulo, M.W.; Hagen, J.J.; Zhou, X.; Guo, Y.; Karten, J.; Pandya, D.; Tilly, T.; et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicate FBLN2, PDGFD and rare de novo variants in PAH. bioRxiv 2020, 2029, 124255. [Google Scholar]
- Zhu, N.; PAH Biobank Enrolling Centers’ Investigators; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; McGoon, M.D.; Elliott, C.G.; Foreman, A.J.; Miller, D.P.; Ivy, D.D. Survival in childhood pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012, 125, 113–122. [Google Scholar] [CrossRef]
- Zhu, N.; Gonzaga-Jauregui, C.; Welch, C.L.; Ma, L.; Qi, H.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; Austin, E.D.; et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ. Genom. Precis. Med. 2018, 11, e001887. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Baker, C.; Gien, J.; Mourani, P.; Galambos, C. The Robyn Barst memorial lecture: Differences between the fetal, newborn, and adult pulmonary circulations: Relevance for age-specific therapies (2013 grover conference series). Pulm. Circ. 2014, 4, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Wynn, J.; Yu, L.; Ms, R.H.; Zhou, X.; Duron, V.; Aspelund, G.; Farkouh-Karoleski, C.; Zygumunt, A.; Krishnan, U.S.; et al. Likely damaging de novo variants in congenital diaphragmatic hernia patients are associated with worse clinical outcomes. Genet. Med. 2020, 1–9. [Google Scholar] [CrossRef]
- Bush, D.; Abman, S.H.; Galambos, C. Prominent intrapulmonary bronchopulmonary anastomoses and abnormal lung development in infants and children with down syndrome. J. Pediatr. 2017, 180, 156–162.e1. [Google Scholar] [CrossRef]
- Sánchez, O.; Dominguez, C.; Ruiz, A.; Ribera, I.; Alijotas-Reig, J.; Roura, L.C.; Carreras, E.; Llurba, E. Angiogenic gene expression in down syndrome fetal hearts. Fetal Diagn. Ther. 2016, 40, 21–27. [Google Scholar] [CrossRef]
- Galambos, C.; Minic, A.D.; Bush, D.; Nguyen, D.; Dodson, B.; Seedorf, G.; Abman, S.H. Increased lung expression of anti-angiogenic factors in down syndrome: Potential role in abnormal lung vascular growth and the risk for pulmonary hypertension. PLoS ONE 2016, 11, e0159005. [Google Scholar] [CrossRef]
- Reuter, M.S.; Jobling, R.; Chaturvedi, R.R.; Manshaei, R.; Costain, G.; Heung, T.; Curtis, M.; Hosseini, S.M.; Liston, E.; Lowther, C.; et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 2018, 21, 1001–1007. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, J.; Shen, J.; Sun, K.; Zhu, J.; Yu, T.; Ji, W.; Chen, Y.; Fu, Q.; Li, F. Mutations in VEGFA are associated with congenital left ventricular outflow tract obstruction. Biochem. Biophys. Res. Commun. 2010, 396, 483–488. [Google Scholar] [CrossRef]
- Beghetti, M.; Rudzinski, A.; Zhang, M. Efficacy and safety of oral sildenafil in children with Down syndrome and pulmonary hypertension. BMC Cardiovasc. Disord. 2017, 17, 177. [Google Scholar] [CrossRef]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 2018, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Byers, H.M.; Dagle, J.M.; Klein, J.M.; Ryckman, K.K.; McDonald, E.L.; Murray, J.C.; Borowski, K.S. Variations in CRHR1 are associated with persistent pulmonary hypertension of the newborn. Pediatr. Res. 2012, 71, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Metzger, R.J.; Papaioannou, V.E. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet. 2012, 8, e1002866. [Google Scholar] [CrossRef]
- Kanai-Azuma, M.; Kanai, Y.; Gad, J.M.; Tajima, Y.; Taya, C.; Kurohmaru, M.; Sanai, Y.; Yonekawa, H.; Yazaki, K.; Tam, P.P.; et al. Depletion of definitive gut endoderm in Sox17-null mutant mice. Development 2002, 129, 2367–2379. [Google Scholar]
- Lilly, A.J.; Lacaud, G.; Kouskoff, V. SOXF transcription factors in cardiovascular development. Semin. Cell Dev. Biol. 2017, 63, 50–57. [Google Scholar] [CrossRef]
- Sheeba, C.J.; Logan, M.P. The roles of T-box genes in vertebrate limb development. Curr. Top Dev. Biol. 2017, 122, 355–381. [Google Scholar] [PubMed]
- Ballif, B.C.; Theisen, A.; Rosenfeld, J.A.; Traylor, R.N.; Gastier-Foster, J.; Thrush, D.L.; Astbury, C.; Bartholomew, D.; McBride, K.L.; Pyatt, R.E.; et al. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am. J. Hum. Genet. 2010, 86, 454–461. [Google Scholar] [CrossRef]
- Nimmakayalu, M.; Sheffield, V.C.; Solomon, D.H.; Patil, S.R.; Shchelochkov, O.A.; Major, H.; Smith, R.J. Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am. J. Med. Genet. Part A 2011, 155A, 418–423. [Google Scholar] [CrossRef]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.H.F.; Roofthooft, M.T.R.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef]
- Levy, M.; Eyries, M.; Szezepanski, I.; Ladouceur, M.; Nadaud, S.; Bonnet, D.; Soubrier, F. Genetic analyses in a cohort of children with pulmonary hypertension. Eur. Respir. J. 2016, 48, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Navas, P.; Tenorio, J.; Quezada, C.A.; Barrios, E.; Gordo, G.; Arias, P.; Messeguer, M.L.; Santos-Lozano, A.; Doza, J.P.; Lapunzina, P.; et al. Molecular analysis of BMPR2, TBX4, and KCNK3 and genotype-phenotype correlations in Spanish patients and families with idiopathic and hereditary pulmonary arterial hypertension. Rev. Esp. Cardiol. 2016, 69, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; Mckean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Galambos, C.; Mullen, M.P.; Shieh, J.T.; Schwerk, N.; Kielt, M.J.; Ullmann, N.; Boldrini, R.; Stucin-Gantar, I.; Haass, C.; Bansal, M.; et al. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and pediatric pulmonary hypertension. Eur. Respir. J. 2019, 54, 1801965. [Google Scholar] [CrossRef] [PubMed]
- Thoré, P.; Girerd, B.; Jaïs, X.; Savale, L.; Ghigna, M.R.; Eyries, M.; Levy, M.; Ovaert, C.; Servettaz, A.; Guillaumot, A.; et al. Phenotype and outcome of pulmonary arterial hypertension patients carrying a TBX4 mutation. Eur. Respir. J. 2020, 55, 1902340. [Google Scholar] [CrossRef]
- Austin, E.D.; Elliott, C.G. TBX4 syndrome: A systemic disease highlighted by pulmonary arterial hypertension in its most severe form. Eur. Respir. J. 2020, 55, 2000585. [Google Scholar] [CrossRef]
- Corada, M.; Orsenigo, F.; Morini, M.F.; Pitulescu, M.E.; Bhat, G.; Nyqvist, D.; Breviario, F.; Conti, V.; Briot, A.; Iruela-Arispe, M.L.; et al. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat. Commun. 2013, 4, 2609. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Hara, K.; Kanai-Azuma, M.; Matsui, T.; Miura, Y.; Tsunekawa, N.; Kurohmaru, M.; Saijoh, Y.; Koopman, P.; Kanai, Y. Redundant roles of Sox17 and Sox18 in early cardiovascular development of mouse embryos. Biochem. Biophys. Res. Commun. 2007, 360, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.W.; Haitchi, H.M.; LeCras, T.D.; Sridharan, A.; Xu, Y.; Wert, S.E.; James, J.; Udell, N.; Thurner, P.J.; Whitsett, J.A. Sox17 is required for normal pulmonary vascular morphogenesis. Dev. Biol. 2014, 387, 109–120. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Marsboom, G.; Jambusaria, A.; Xiong, S.; Toth, P.T.; Benevolenskaya, E.V.; Rehman, J.; Malik, A.B. Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat. Commun. 2019, 10, 2126. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Kataoka, M.; Suzuki, H.; Aimi, Y.; Chiba, T.; Kanekura, K.; Satoh, T.; Fukuda, K.; Gamou, S.; Kosaki, K. SOX17 Mutations in Japanese patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 1231–1233. [Google Scholar] [CrossRef]
- Liu, X.; Luo, M.; Xie, W.; Wells, J.M.; Goodheart, M.J.; Engelhardt, J.F. Sox17 modulates Wnt3A/β-catenin-mediated transcriptional activation of the Lef-1 promoter. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L694–L710. [Google Scholar] [CrossRef]
- Banerjee, A.; Ray, S. Structural insight, mutation and interactions in human β-catenin and SOX17 protein: A molecular-level outlook for organogenesis. Gene 2017, 610, 118–126. [Google Scholar] [CrossRef]
- Zhang, H.S.; Liu, Q.; Piao, C.M.; Zhu, Y.; Li, Q.Q.; Du, J.; Gu, H. Genotypes and phenotypes of Chinese pediatric patients with idiopathic and heritable pulmonary arterial hypertension—A single-center study. Can. J. Cardiol. 2019, 35, 1851–1856. [Google Scholar] [CrossRef]
- Haarman, M.G.; Kerstjens-Frederikse, W.S.; Vissia-Kazemier, T.R.; Breeman, K.T.; Timens, W.; Vos, Y.J.; Roofthooft, M.T.; Hillege, H.L.; Berger, R.M. The genetic epidemiology of pediatric pulmonary arterial hypertension. J. Pediatr. 2020, 225, 65–73. [Google Scholar] [CrossRef]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef]
- Qi, H.; Yu, L.; Zhou, X.; Wynn, J.; Zhao, H.; Guo, Y.; Zhu, N.; Kitaygorodsky, A.; Hernan, R.; Aspelund, G.; et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018, 14, e1007822. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef]
- Epi, K.C.; Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef]
- Tartaglia, M.; Martinelli, S.; Stella, L.; Bocchinfuso, G.; Flex, E.; Cordeddu, V.; Zampino, G.; Van Der Burgt, I.; Palleschi, A.; Petrucci, T.C.; et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am. J. Hum. Genet. 2006, 78, 279–290. [Google Scholar] [CrossRef]
- Hopper, R.K.; Feinstein, J.A.; Manning, M.A.; Benitz, W.; Hudgins, L. Neonatal pulmonary arterial hypertension and Noonan syndrome: Two fatal cases with a specific RAF1 mutation. Am. J. Med. Genet. Part. A 2015, 167A, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Tinker, A.; Uren, N.; Schofield, J. Severe pulmonary hypertension in Ullrich-Noonan syndrome. Br. Heart J. 1989, 62, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Troyanovsky, B.; Levchenko, T.; Mansson, G.; Matvijenko, O.; Holmgren, L. Angiomotin: An angiostatin binding protein that regulates endothelial cell migration and tube formation. J. Cell Biol. 2001, 152, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, L.; Ambrosino, E.; Birot, O.; Tullus, C.; Veitonmaki, N.; Levchenko, T.; Carlson, L.M.; Musiani, P.; Iezzi, M.; Curcio, C.; et al. A DNA vaccine targeting angiomotin inhibits angiogenesis and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2006, 103, 9208–9213. [Google Scholar] [CrossRef]
- Zheng, Y.; Vertuani, S.; Nyström, S.; Audebert, S.; Meijer, I.; Tegnebratt, T.; Borg, J.P.; Uhlén, P.; Majumdar, A.; Holmgren, L. Angiomotin-like protein 1 controls endothelial polarity and junction stability during sprouting angiogenesis. Circ. Res. 2009, 105, 260–270. [Google Scholar] [CrossRef]
- Mohamed, B.A.; Barakat, A.Z.; Held, T.; Elkenani, M.; Mühlfeld, C.; Männer, J.; Adham, I.M. Respiratory distress and early neonatal lethality in Hspa4l/Hspa4 double-mutant mice. Am. J. Respir. Cell Mol. Biol. 2014, 50, 817–824. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; De Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Gong, J.; Thimmulappa, R.K.; Kosmider, B.; Biswal, S.; Duh, E.J. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc. Natl. Acad. Sci. USA 2013, 110, E3910–E3918. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and its activators in respiratory diseases. Oxid. Med. Cell. Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef]
- Samocha, K.E.; Robinson, E.B.; Sanders, S.J.; Stevens, C.; Sabo, A.; McGrath, L.M.; Kosmicki, J.A.; Rehnström, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Chida, A.; Shintani, M.; Yagi, H.; Fujiwara, M.; Kojima, Y.; Sato, H.; Imamura, S.; Yokozawa, M.; Onodera, N.; Horigome, H.; et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am. J. Cardiol. 2012, 110, 586–593. [Google Scholar] [CrossRef]
- Wang, X.J.; Lian, T.Y.; Jiang, X.; Liu, S.F.; Li, S.Q.; Jiang, R.; Wu, W.H.; Ye, J.; Cheng, C.Y.; Du, Y.; et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nat. Cell Biol. 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Hemnes, A.R. Using omics to understand and treat pulmonary vascular disease. Front. Med. 2018, 5, 157. [Google Scholar] [CrossRef]
- Harbaum, L.; Rhodes, C.J.; Otero-Núñez, P.; Wharton, J.; Wilkins, M.R. The application of ‘omics’ to pulmonary arterial hypertension. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Cogan, J.; Geraci, M.W.; Robinson, L.; Newman, J.H.; Phillips, J.A.; Lane, K.B.; Meyrick, B.; Loyd, J.E. Gene expression in BMPR2 mutation carriers with and without evidence of Pulmonary Arterial Hypertension suggests pathways relevant to disease penetrance. BMC Med. Genom. 2008, 1, 45. [Google Scholar] [CrossRef] [PubMed]
- Prosseda, S.D.; Tian, X.; Kuramoto, K.; Boehm, M.; Sudheendra, D.; Miyagawa, K.; Zhang, F.; Solow-Cordero, D.; Saldivar, J.C.; Austin, E.D.; et al. FHIT, a novel modifier gene in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 83–98. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Ghataorhe, P.; Wharton, J.; Rue-Albrecht, K.C.; Hadinnapola, C.; Watson, G.; Bleda, M.; Haimel, M.; Coghlan, G.; Corris, P.A.; et al. Plasma metabolomics implicates modified transfer RNAs and altered bioenergetics in the outcomes of pulmonary arterial hypertension. Circulation 2017, 135, 460–475. [Google Scholar] [CrossRef]
- Stearman, R.S.; Bui, Q.M.; Speyer, G.; Handen, A.; Cornelius, A.R.; Graham, B.B.; Kim, S.; Mickler, E.A.; Tuder, R.M.; Chan, S.Y.; et al. Systems analysis of the human pulmonary arterial hypertension lung transcriptome. Am. J. Respir. Cell Mol. Biol. 2019, 60, 637–649. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Trammell, A.W.; Archer, S.L.; Rich, S.; Yu, C.; Nian, H.; Penner, N.; Funke, M.; Wheeler, L.; Robbins, I.M.; et al. Peripheral blood signature of vasodilator-responsive pulmonary arterial hypertension. Circulation 2014, 131, 401–409. [Google Scholar] [CrossRef]
- Abman, S.H.; Raj, U. Towards improving the care of children with pulmonary hypertension: The rationale for developing a pediatric pulmonary hypertension network. Prog. Pediatr. Cardiol. 2009, 27, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Kinsella, J.P.; Rosenzweig, E.B.; Krishnan, U.; Kulik, T.; Mullen, M.; Wessel, D.L.; Steinhorn, R.; Adatia, I.; Hanna, B.D.; et al. Implications of the U.S. Food and drug administration warning against the use of sildenafil for the treatment of pediatric pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, U.; Feinstein, J.A.; Adatia, I.; Austin, E.D.; Mullen, M.P.; Hopper, R.K.; Hanna, B.D.; Romer, L.; Keller, R.L.; Fineman, J.; et al. Evaluation and management of pulmonary hypertension in children with bronchopulmonary dysplasia. J. Pediatr. 2017, 188, 24–34.E1. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, J.P.; Steinhorn, R.; Mullen, M.P.; Hopper, R.K.; Keller, R.L.; Ivy, D.D.; Austin, E.D.; Krishnan, U.; Rosenzweig, E.B.; Fineman, J.R.; et al. The left ventricle in congenital diaphragmatic hernia: Implications for the management of pulmonary hypertension. J. Pediatr. 2018, 197, 17–22. [Google Scholar] [CrossRef]
- Levy, P.T.; Jain, A.; Nawaytou, H.; Teitel, D.; Keller, R.; Fineman, J.; Steinhorn, R.; Abman, S.H.; McNamara, P.J. Risk assessment and monitoring of chronic pulmonary hypertension in premature infants. J. Pediatr. 2020, 217, 199–209.e4. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Beck, G.J.; Newman, J.H.; Abidov, A.; Aldred, M.A.; Barnard, J.; Berman, R.E.; Borlaug, B.A.; Chung, W.K.; Comhair, S.A.A.; et al. PVDOMICS: A multi-center study to improve understanding of pulmonary vascular disease through phenomics. Circ. Res. 2017, 121, 1136–1139. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Del Cerro, M.J.; Abman, S.; Diaz, G.; Freudenthal, A.H.; Freudenthal, F.; Harikrishnan, S.; Haworth, S.G.; Ivy, D.; Lopes, A.A.; Raj, J.U.; et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI pediatric taskforce, Panama 2011. Pulm. Circ. 2011, 1, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, P.; Daniels, A.M.; Snyder, L.G.; Beaumont, A.; Camba, A.; Esler, A.; Gulsrud, A.; Mason, A.; Gutierrez, A.; Nicholson, A.; et al. SPARK: A US cohort of 50,000 families to accelerate autism research. Neuron 2018, 97, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, P.; The SPARK Consortium; Zhou, X.; Astrovskaya, I.; Turner, T.N.; Wang, T.; Brueggeman, L.; Barnard, R.; Hsieh, A.; Snyder, L.G.; et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom. Med. 2019, 4, 19. [Google Scholar] [CrossRef]
- Chaisson, N.F.; Dodson, M.W.; Elliott, C.G. Pulmonary capillary hemangiomatosis and pulmonary veno-occlusive disease. Clin. Chest Med. 2016, 37, 523–534. [Google Scholar] [CrossRef]
- Plauchu, H.; De Chadarévian, J.P.; Bideau, A.; Robert, J.-M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am. J. Med. Genet. 1989, 32, 291–297. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group (n) | Age at dx (y) | F:M Ratio | mPAP (mm Hg) | mPCWP (mm Hg) | CO Fisk (L/min) | PVR (Woods Units) | Common Comorbidities |
|---|---|---|---|---|---|---|---|
| Child (226) | 7.7 ± 5.4 | 1.65:1 | 55.1 ± 18.6 | 9.0 ± 3.0 | 3.2 ± 1.6 | 18.1 ± 11.7 | CHD, CDH, DS, lung growth/development |
| Adult (2345) | 51.6 ± 14.7 | 4.02:1 | 49.6 ± 13.9 | 10.2 ± 4.2 | 4.6 ± 1.7 | 10.0 ± 5.9 | HTN, hypothyroidism, other pulmonary & metabolic diseases |
| p-value | <0.0001 * | <0.0001 ** | <0.0001 * | <0.0001 * | <0.0001 * | <0.0001 * |
| Term | Reactome ID | # Genes in Overlap | p-Value | Adjusted p-Value | Genes |
|---|---|---|---|---|---|
| Developmental biology | R-HAS-1266738 | 16/786 | 6.8 × 10−5 | 0.03 | KLB, ROBO2, LAMA1, EGF, ANK3, LAMC, SLC2A4, MED6, SPRED2, MEIS1, NRFA2, PCMC4, NF1, EP300, TCF4, EPHB4 |
| Transmembrane transport of small molecules | R-HAS-382561 | 13/594 | 1.7 ×10−4 | 0.03 | RYR2, ABCC4, ABCC1, SLC1A3, SL#3A4, SLC8A1, CLCN5, SLCA9, ATPB7, ASPH, WNK1, NUP35, EMB |
| Non-integrin membrane extracellular matrix interactions | R-HAS-3000171 | 4/42 | 1.7 × 10−4 | 0.03 | LAMA1, LAMA4, LAMC1, THBS1 |
| Ion homeostasis | R-HAS-5578775 | 4/51 | 1.7 × 10−4 | 0.03 | RYR2, ASPH, TPR3, SLC8A1 |
| Variant Type * | Observed in Trios (n = 124) | Expected by Chance | Enrichment | p-Value | Estimated # of True Risk Variants |
|---|---|---|---|---|---|
| SYN | 18 | 14.0 | 1.3 | 0.28 | --- |
| LGD | 11 | 4.7 | 2.4 | 0.06 | --- |
| MIS | 40 | 31.7 | 1.3 | 0.15 | --- |
| D-MIS | 19 | 7.2 | 2.6 | 2.0 × 10−4 | 12 |
| LGD + D-MIS | 30 | 11.8 | 2.5 | 7.0 × 10−6 | 18 |
| Gene Symbol | Variant Type | Protein Change | REVEL Score | CADD Score | Allele Frequency (gnomAD) | E16.5 Lung Expression Rank | E14.5 Heart Expression Rank | Variant Carrier PAH Subtype |
|---|---|---|---|---|---|---|---|---|
| AMOT | LGD | p.(Leu320Cysfs*55) | . | 31 | . | 68 | 95 | IPAH |
| CSNK2A2 | D-MIS | p.(His184Leu) | 0.50 | 25 | . | 55 | 77 | IPAH |
| HNRNPF | LGD | p.(Tyr210Leufs*14) | . | 29 | . | 85 | 98 | PPHN, PAH |
| HSPA4 | D-MIS | p.(pro684Arg) | 0.62 | 30 | 4.1 × 10−6 | 43 | 96 | PAH-CHD |
| KDM3B | D-MIS | p.(Pro1100Ser) | 0.66 | 29 | . | 89 | 87 | IPAH |
| KEAP1 | LGD | p.(Tyr584*) | . | 35 | . | 79 | 82 | IPAH with dev delay |
| MECOM | D-MIS | p.(Phe762Ser) | 0.76 | 32 | . | 82 | 60 | IPAH |
| ZMYM2 | LGD | p.(Arg540*) | . | 36 | . | 93 | 77 | IPAH with skeletal anomalies |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welch, C.L.; Chung, W.K. Genetics and Genomics of Pediatric Pulmonary Arterial Hypertension. Genes 2020, 11, 1213. https://doi.org/10.3390/genes11101213
Welch CL, Chung WK. Genetics and Genomics of Pediatric Pulmonary Arterial Hypertension. Genes. 2020; 11(10):1213. https://doi.org/10.3390/genes11101213
Chicago/Turabian StyleWelch, Carrie L., and Wendy K. Chung. 2020. "Genetics and Genomics of Pediatric Pulmonary Arterial Hypertension" Genes 11, no. 10: 1213. https://doi.org/10.3390/genes11101213
APA StyleWelch, C. L., & Chung, W. K. (2020). Genetics and Genomics of Pediatric Pulmonary Arterial Hypertension. Genes, 11(10), 1213. https://doi.org/10.3390/genes11101213