BMPR2 Promoter Variants Effect Gene Expression in Pulmonary Arterial Hypertension Patients

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Genetic Analysis
2.3. Plasmid Construction
2.4. Cell Culture and Luciferase Assay
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristics
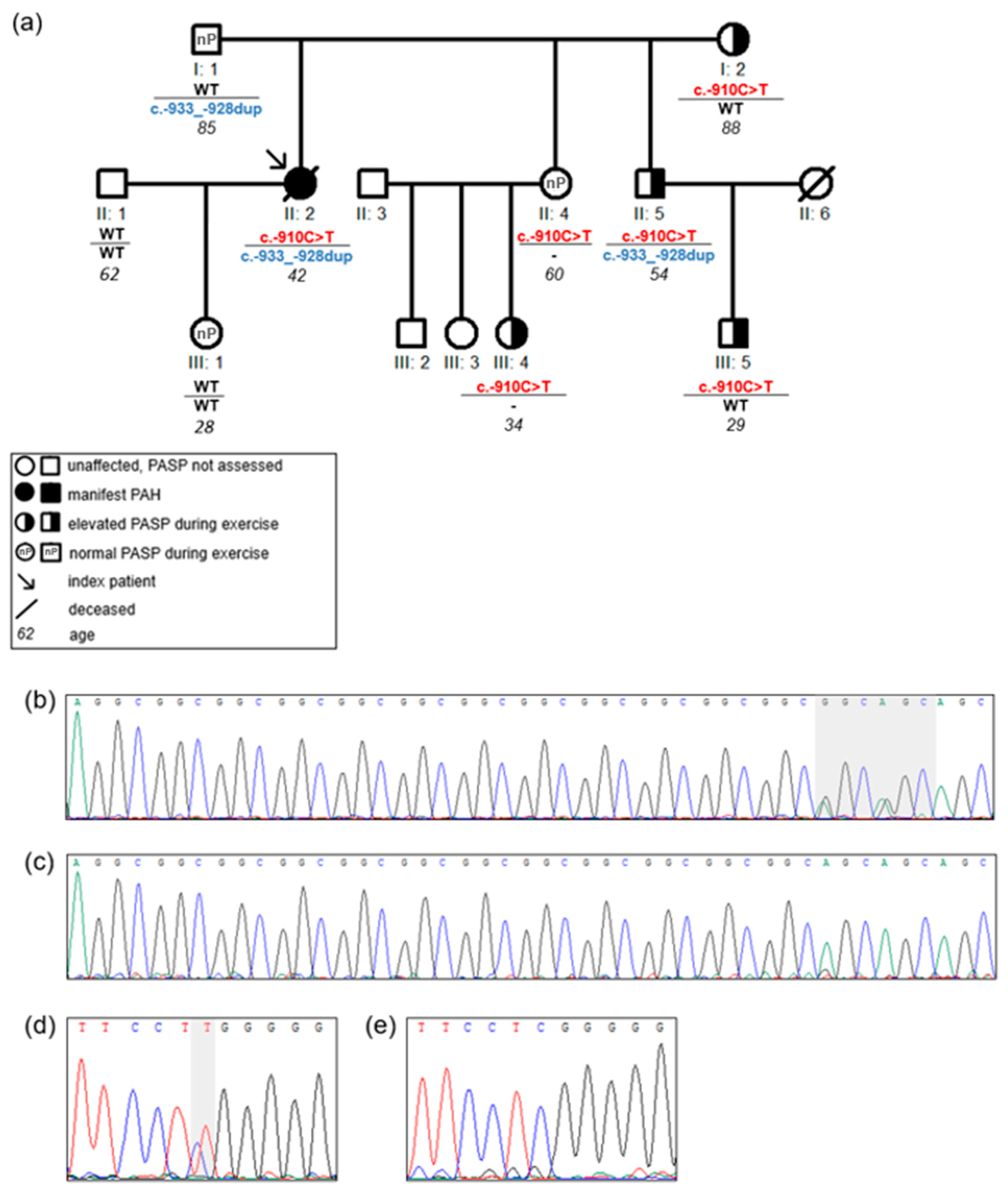
3.2. Genetic Analysis
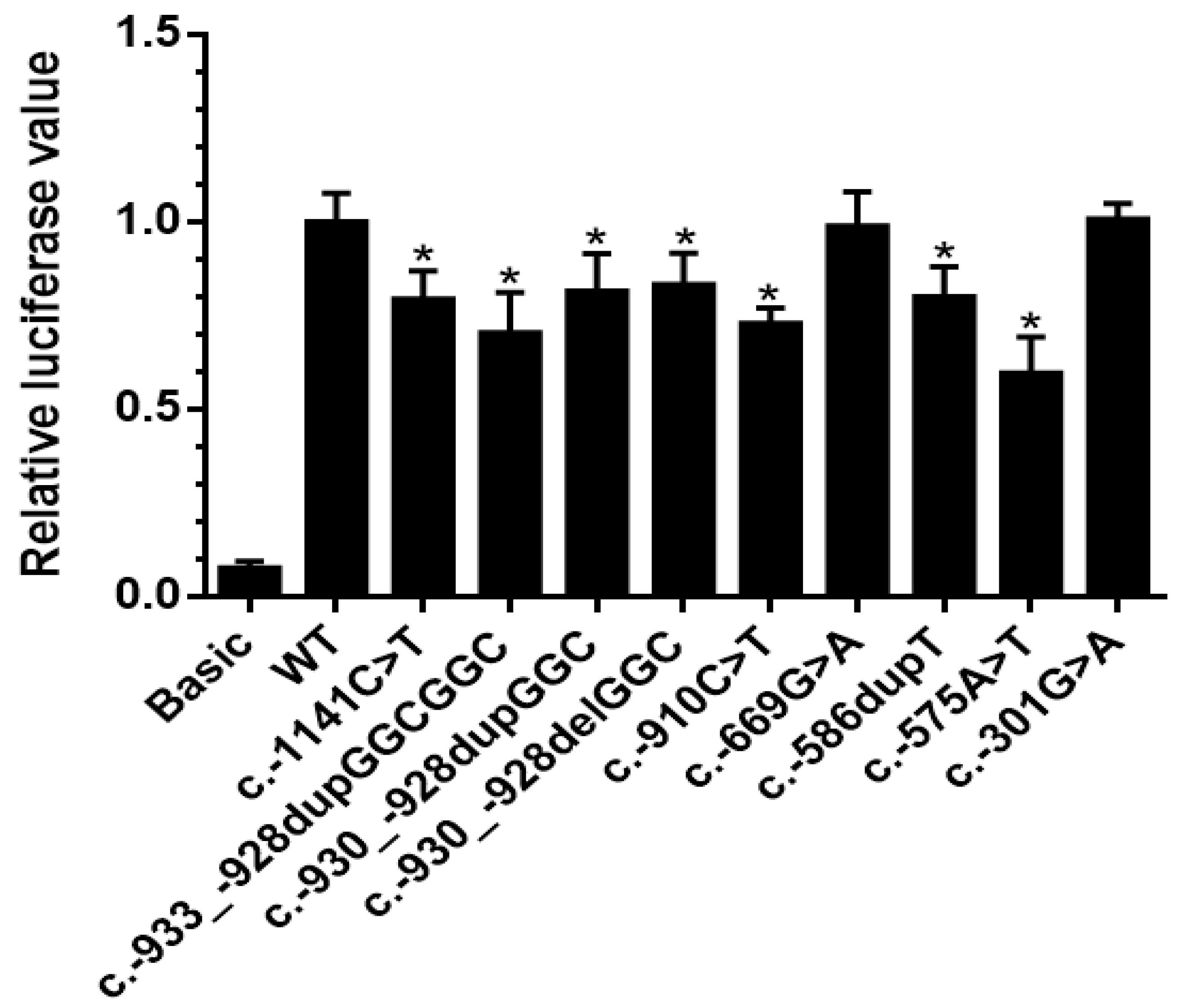
3.3. Effect of Promoter Variants on Gene Expression
3.4. Association of Promoter Variants with an Abnormal Pulmonary Artery Pressure during Exercise or PAH Manifestation
4. Discussion
4.1. Impact of Promoter Variants on Transcription Factor Binding Sites
4.2. Contradictions to Previous Studies
4.3. Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiéry, J.-L.; Gibbs, S.; Lang, I.M.; Kaminski, K.A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Hear. J. 2015, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Pfarr, N.; Szamalek-Hoegel, J.; Fischer, C.; Hinderhofer, K.; Nagel, C.; Ehlken, N.; Tiede, H.; Olschewski, H.; Reichenberger, F.; Ghofrani, H.A.; et al. Hemodynamic and clinical onset in patients with hereditary pulmonary arterial hypertension and BMPR2 mutations. Respir. Res. 2011, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Berghausen, E.M.; Eichstaedt, C.A.; Fleischmann, B.K.; Grünig, E.; Grünig, G.; Hansmann, G.; Harbaum, L.; Hennigs, J.K.; Jonigk, D.; et al. Pathobiology, pathology and genetics of pulmonary hypertension: Update from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272, 4–10. [Google Scholar] [CrossRef]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galié, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.; Phillips, J.A.; Hamid, R.; Loyd, J.E. Longitudinal Analysis Casts Doubt on the Presence of Genetic Anticipation in Heritable Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef]
- Frydman, N.A.; Steffann, J.; Girerd, B.; Frydman, R.; Munnich, A.; Simonneau, G.; Humbert, M. Pre-implantation genetic diagnosis in pulmonary arterial hypertension due to BMPR2 mutation. Eur. Respir. J. 2012, 39, 1534–1535. [Google Scholar] [CrossRef]
- Eichstaedt, C.A.; Song, J.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grünig, E.; Hinderhofer, K. EIF2AK4 mutation as “second hit” in hereditary pulmonary arterial hypertension. Respir. Res. 2016, 17, 1–8. [Google Scholar] [CrossRef]
- Wang, G.; Knight, L.; Ji, R.; Lawrence, P.; Kanaan, U.; Li, L.; Das, A.; Cui, B.; Zou, W.; Penny, D.J.; et al. Early onset severe pulmonary arterial hypertension with ‘two-hit’ digenic mutations in both BMPR2 and KCNA5 genes. Int. J. Cardiol. 2014, 177, e167–e169. [Google Scholar] [CrossRef]
- Viales, R.R.; Eichstaedt, C.A.; Ehlken, N.; Fischer, C.; Lichtblau, M.; Grünig, E.; Hinderhofer, K. Mutation in BMPR2 Promoter: A ‘Second Hit’ for Manifestation of Pulmonary Arterial Hypertension? PLoS ONE 2015, 10, e0133042. [Google Scholar] [CrossRef] [PubMed]
- Hinderhofer, K.; Fischer, C.; Pfarr, N.; Szamalek-Hoegel, J.; Lichtblau, M.; Nagel, C.; Egenlauf, B.; Ehlken, N.; Grünig, E. Identification of a New Intronic BMPR2-Mutation and Early Diagnosis of Heritable Pulmonary Arterial Hypertension in a Large Family with Mean Clinical Follow-up of 12 Years. PLoS ONE 2014, 9, e91374. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.; Machado, R.D.; James, V.; Morrell, N.W.; Trembath, R. Characterization of theBMPR25′-Untranslated Region and a Novel Mutation in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2007, 176, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, W.; Zhang, W.; Sun, K.; Song, X.; Gao, S.; Zhang, C.; Hui, R.; Hu, H. Novel promoter and exon mutations of the BMPR2 gene in Chinese patients with pulmonary arterial hypertension. Eur. J. Hum. Genet. 2009, 17, 1063–1069. [Google Scholar] [CrossRef]
- Pousada, G.; Lupo, V.; Cástro-Sánchez, S.; Álvarez-Satta, M.; Sánchez-Monteagudo, A.; Baloira, A.; Espinós, C.; Valverde, D. Molecular and functional characterization of the BMPR2 gene in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 1923. [Google Scholar] [CrossRef]
- Limsuwan, A.; Choubtum, L.; Wattanasirichaigoon, D. 5′UTR Repeat Polymorphisms of the BMPR2 gene in Children with Pulmonary Hypertension associated with Congenital Heart Disease. Hear. Lung Circ. 2013, 22, 204–210. [Google Scholar] [CrossRef]
- Galiè, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.-L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Hear. J. 2009, 30, 2493–2537. [Google Scholar] [CrossRef]
- Nagel, C.; Henn, P.; Ehlken, N.; D’Andrea, A.; Blank, P.D.N.; Bossone, E.; Böttger, A.; Fiehn, C.; Fischer, C.; Lorenz, H.-M.; et al. Stress Doppler echocardiography for early detection of systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Res. 2015, 17, 165. [Google Scholar] [CrossRef]
- Grünig, E.; Janssen, B.; Mereles, D.; Barth, U.; Borst, M.M.; Vogt, I.R.; Fischer, C.; Olschewski, H.; Kuecherer, H.F.; Kübler, W. Abnormal Pulmonary Artery Pressure Response in Asymptomatic Carriers of Primary Pulmonary Hypertension Gene. Circulation 2000, 102, 1145–1150. [Google Scholar] [CrossRef]
- Song, J.; Eichstaedt, C.A.; Viales, R.R.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grünig, E.; Hinderhofer, K. Identification of genetic defects in pulmonary arterial hypertension by a new gene panel diagnostic tool. Clin. Sci. 2016, 130, 2043–2052. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, C.; Liu, C.; Wang, W.; Zhang, N.; Hadadi, C.; Huang, J.; Zhong, N.; Lu, W. Functional mutations in 5′UTR of the BMPR2 gene identified in Chinese families with pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Nakanishi, G.; Lewandoski, M.; Jetten, A.M. GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 2003, 31, 5513–5525. [Google Scholar] [CrossRef] [PubMed]
- Eichstaedt, C.A.; Song, J.; Viales, R.R.; Pan, Z.; Benjamin, N.; Fischer, C.; Hoeper, M.; Ulrich, S.; Hinderhofer, K.; Grünig, E. First identification of Krüppel-like factor 2 mutation in heritable pulmonary arterial hypertension. Clin. Sci. 2017, 131, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Sindi, H.A.; Russomanno, G.; Satta, S.; Abdul-Salam, V.B.; Jo, K.B.; Qazi-Chaudhry, B.; Ainscough, A.J.; Szulcek, R.; Bogaard, H.J.; Morgan, C.C.; et al. Author Correction: Therapeutic potential of KLF2-induced exosomal microRNAs in pulmonary hypertension. Nat. Commun. 2020, 11, 1. [Google Scholar] [CrossRef]
- Voz, M.L.; Agten, N.S.; Van De Ven, W.J.; Kas, K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000, 60, 106–113. [Google Scholar]
- Nishi, H.; Nishi, K.H.; Johnson, A.C. Early Growth Response-1 gene mediates up-regulation of epidermal growth factor receptor expression during hypoxia. Cancer Res. 2002, 62, 827–834. [Google Scholar]
- Gaddipati, R.; West, J.; Loyd, J.E.; Blackwell, T.; Lane, K.A. EGR1 is essential for transcriptional regulation of BMPR2. Am. J. Mol. Biol. 2011, 1, 131–139. [Google Scholar] [CrossRef][Green Version]
- Lopes, M.; Goupille, O.; Cloment, C.S.; Lallemand, Y.; Cumano, A.; Robert, B. Msx genes define a population of mural cell precursors required for head blood vessel maturation. Development 2011, 138, 3055–3066. [Google Scholar] [CrossRef]
- GTExPortal Database Release V8. Available online: www.gtexportal.org (accessed on 8 March 2020).




{kind=link}
{kind=link}
{kind=link}
| Characteristic * | Mean ± SD | Min | Max |
|---|---|---|---|
| Women [%] | 43 | ||
| Age at diagnosis [years] | 34.0 ± 14.2 | 13 | 56 |
| Heart rate [min−1] | 88.6 ± 12.2 | 68 | 99 |
| Oxygen saturation [%] | 94.0 ± 5.0 | 87 | 98 |
| Mean pulmonary artery pressure [mmHg] | 60.3 ± 9.4 | 46 | 70 |
| Pulmonary arterial wedge pressure [mmHg] | 5.0 ± 1.6 | 3 | 7 |
| Pulmonary vascular resistance [Wood Units] | 20.2 ± 5.7 | 13 | 26 |
| Cardiac index [ml/min/m2] | 2.1 ± 0.9 | 1.3 | 3.6 |
| Nucleotide Change | Patients | Variant Carriers | GnomAD Frequency | rsID | Described in PAH Patients |
|---|---|---|---|---|---|
| c.-301G>A 1 | 2 HPAH families | 2 indices 6 family members 2 | 0.69% | rs116154690 | 1 SSc-APAH [15] |
| c.-575A>T | 1 HPAH family | 1 index | 0.04% | rs550462760 | This study |
| c.-586dupT 3 | 1 IPAH patient | 1 index | 0.17% | rs572725320 | This study |
| c.-669G>A | 3 HPAH families | 2 indices 9 family members | 0.95% | rs115604088 | [11,14] |
| c.-910C>T 4 | 1 HPAH family | 1 index 5 family members | - | - | This study |
| c.-930_-928dupGGC (13 repeats) | 1 IPAH patient | 1 index | - | rs375624016 | 4 CHD-APAH and 10 controls; 1 HPAH [16,21] |
| c.-933_-928dupGGCGGC (14 repeats) 4 | 1 HPAH family | 1 index 2 family members | - | - | 1 CHD-APAH [16] |
| c.-930_-928delGGC (11 repeats) | 1 IPAH patient | 1 index | - | rs886055459 | 1 CHD-APAH [16] |
| c.-1141C>T 1 | 1 HPAH family | 1 index 2 family members | - | - | This study |
| Family−/− Index | Other Pathogenic Variants/VUS | BMPR2 Promoter Variants | Gene Expression Compared to Wild-Type | Variant in FM with ↑ Exercise PASP/all FM with ↑ Exercise PASP | Variant in FM with Normal Exercise PASP/All FM with Normal Exercise PASP |
|---|---|---|---|---|---|
| Family 1 | none | c.-910C>T | 0.73 x | 4/4 | 1/2 |
| c.-933_-928 dupGGCGGC | 0.70 x | 1/4 | 1/2 | ||
| Family 2 | EIF2AK4 c.641delA p.(Lys214Argfs*21) | c.-1141C>T | 0.79 x | in index patient | NA |
| c.-301G>A | 1.01 x | in 2nd PAH patient | NA | ||
| Family 3 | none | c.-575A>T | 0.60 x | NA | not in 2nd PAH patient |
| Family 4 | none | c.-301G>A | 1.01 x | 1/2 | 1/1 |
| Family 5 1 | BMPR2 c.244C>T p.(Gln82*) | c.-669G>A | 0.99 x | 3/6 | 0/2 |
| Family 6 | BMPR2 exon 2–3 deletion | c.-669G>A | 0.99 x | 4/6 | 0/2 |
| Family 7 | ENG c.1633G>A p.(Gly545Ser) | c.-669G>A | 0.99 x | 1/1 | 2/5 |
| IPAH 1 | none | c.-586dupT | 0.80 x | NA | NA |
| IPAH 2 | BMPR2 c.1453G>A p.(Asp485Asn) | c.-930_-928delGGC | 0.83 x | NA | NA |
| IPAH 3 | BMPR2 c.2695C>T p.(Arg899*) | c.-930_-928dupGGC | 0.82 x | NA | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Hinderhofer, K.; Kaufmann, L.T.; Benjamin, N.; Fischer, C.; Grünig, E.; Eichstaedt, C.A. BMPR2 Promoter Variants Effect Gene Expression in Pulmonary Arterial Hypertension Patients. Genes 2020, 11, 1168. https://doi.org/10.3390/genes11101168
Song J, Hinderhofer K, Kaufmann LT, Benjamin N, Fischer C, Grünig E, Eichstaedt CA. BMPR2 Promoter Variants Effect Gene Expression in Pulmonary Arterial Hypertension Patients. Genes. 2020; 11(10):1168. https://doi.org/10.3390/genes11101168
Chicago/Turabian StyleSong, Jie, Katrin Hinderhofer, Lilian T. Kaufmann, Nicola Benjamin, Christine Fischer, Ekkehard Grünig, and Christina A. Eichstaedt. 2020. "BMPR2 Promoter Variants Effect Gene Expression in Pulmonary Arterial Hypertension Patients" Genes 11, no. 10: 1168. https://doi.org/10.3390/genes11101168
APA StyleSong, J., Hinderhofer, K., Kaufmann, L. T., Benjamin, N., Fischer, C., Grünig, E., & Eichstaedt, C. A. (2020). BMPR2 Promoter Variants Effect Gene Expression in Pulmonary Arterial Hypertension Patients. Genes, 11(10), 1168. https://doi.org/10.3390/genes11101168