A Pan-Genome Guided Metabolic Network Reconstruction of Five Propionibacterium Species Reveals Extensive Metabolic Diversity


 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Gene Annotation
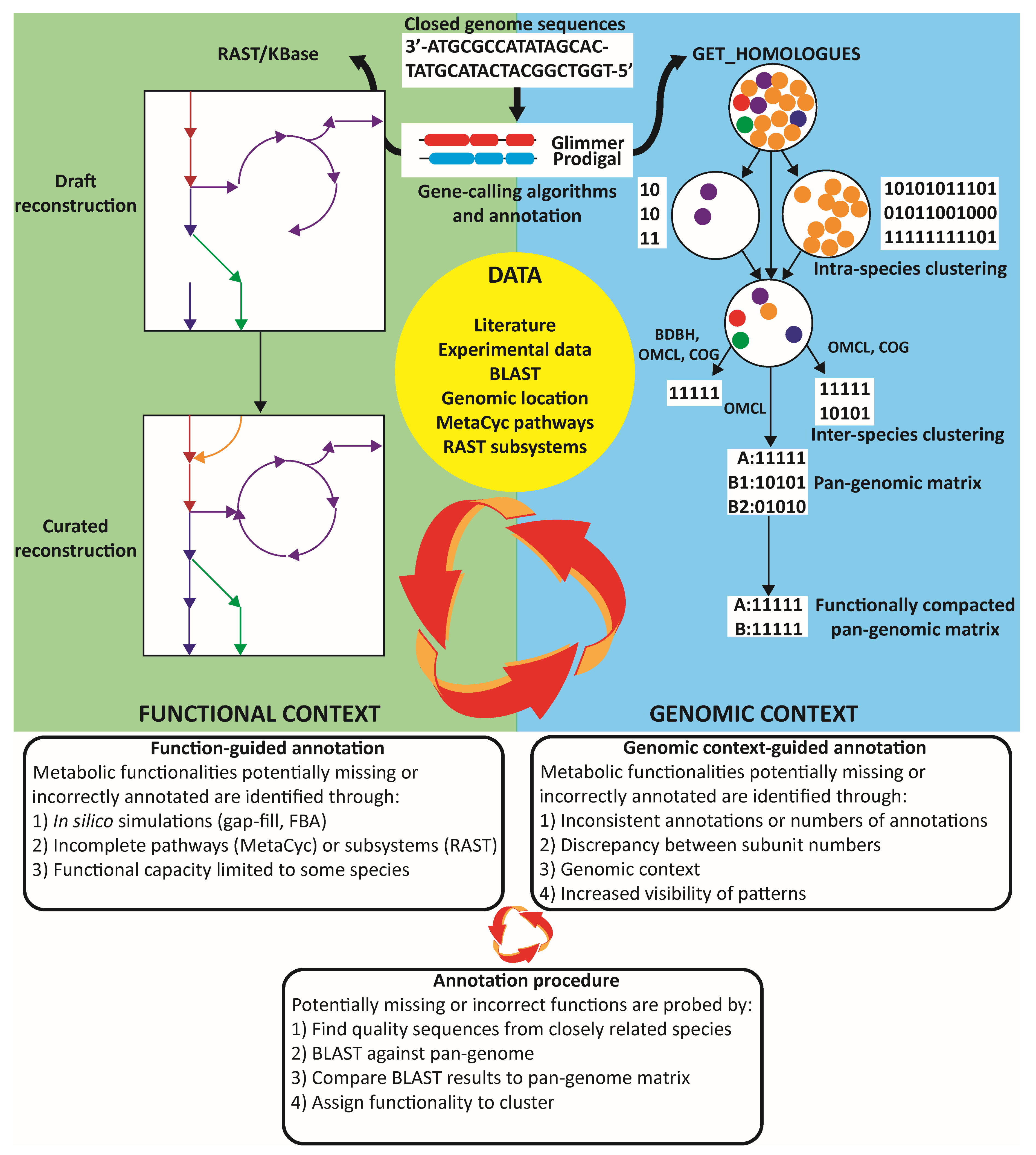
2.2. Pan-Genome Comparisons and Matrix Generation
2.3. Genome-Scale Metabolic Model and Pan-GEM Construction
2.4. Pan-Genome Guided Genome-Scale Metabolic Reconstruction
2.5. Phenotype Array Data Generation and Integration
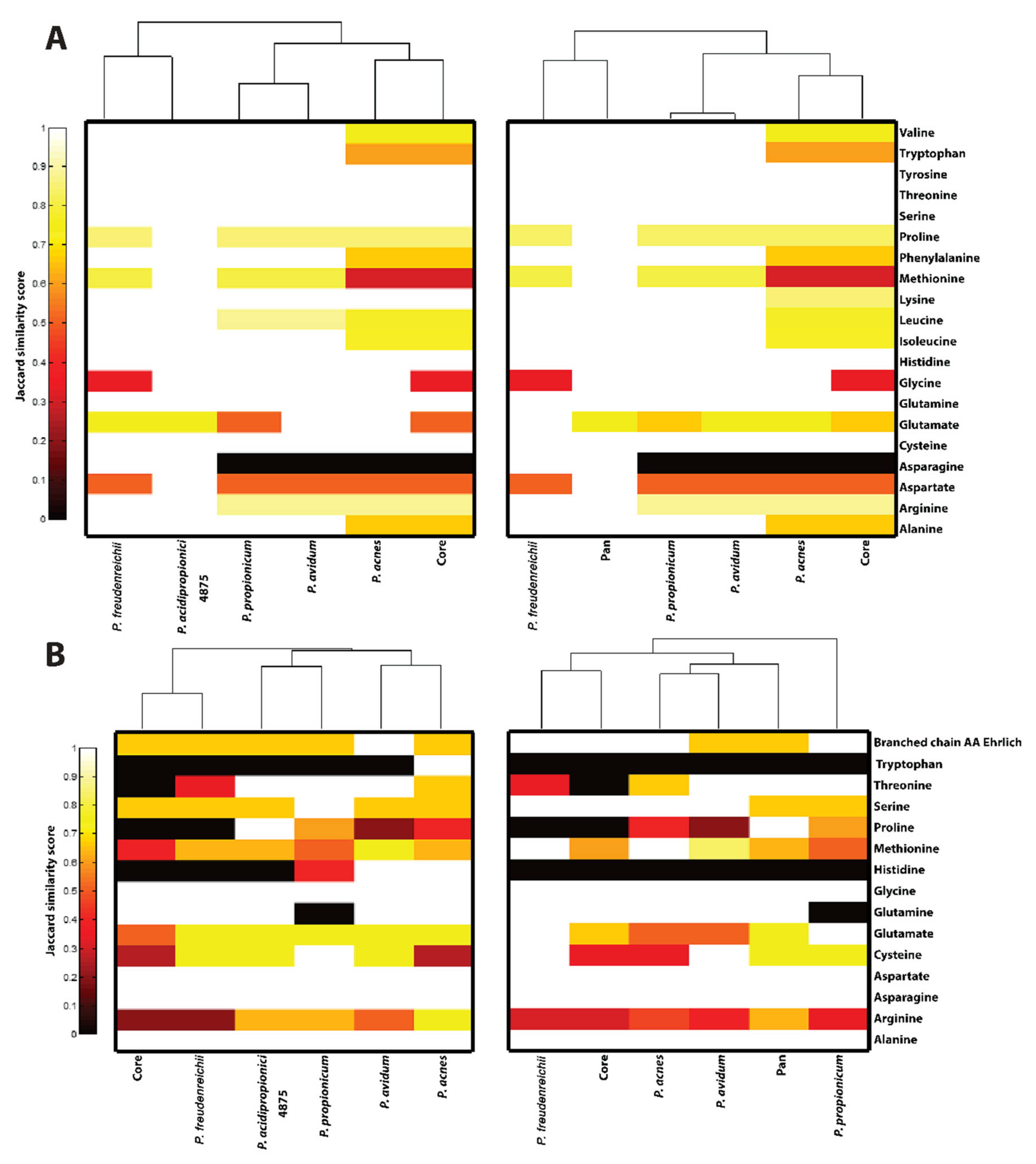
2.6. Clustergram Generation
2.7. Bioreactor Cultures and Omics Analysis
2.8. PFOR Knockout
3. Results and Discussion
3.1. Exploration of Core and Pan-Genome Size
3.2. Proteomics and Transcriptomics of P. acidipropionici
3.3. Model Modifications Summary
3.4. The Metabolism of Propionibacteria
3.4.1. Central Carbon Metabolism
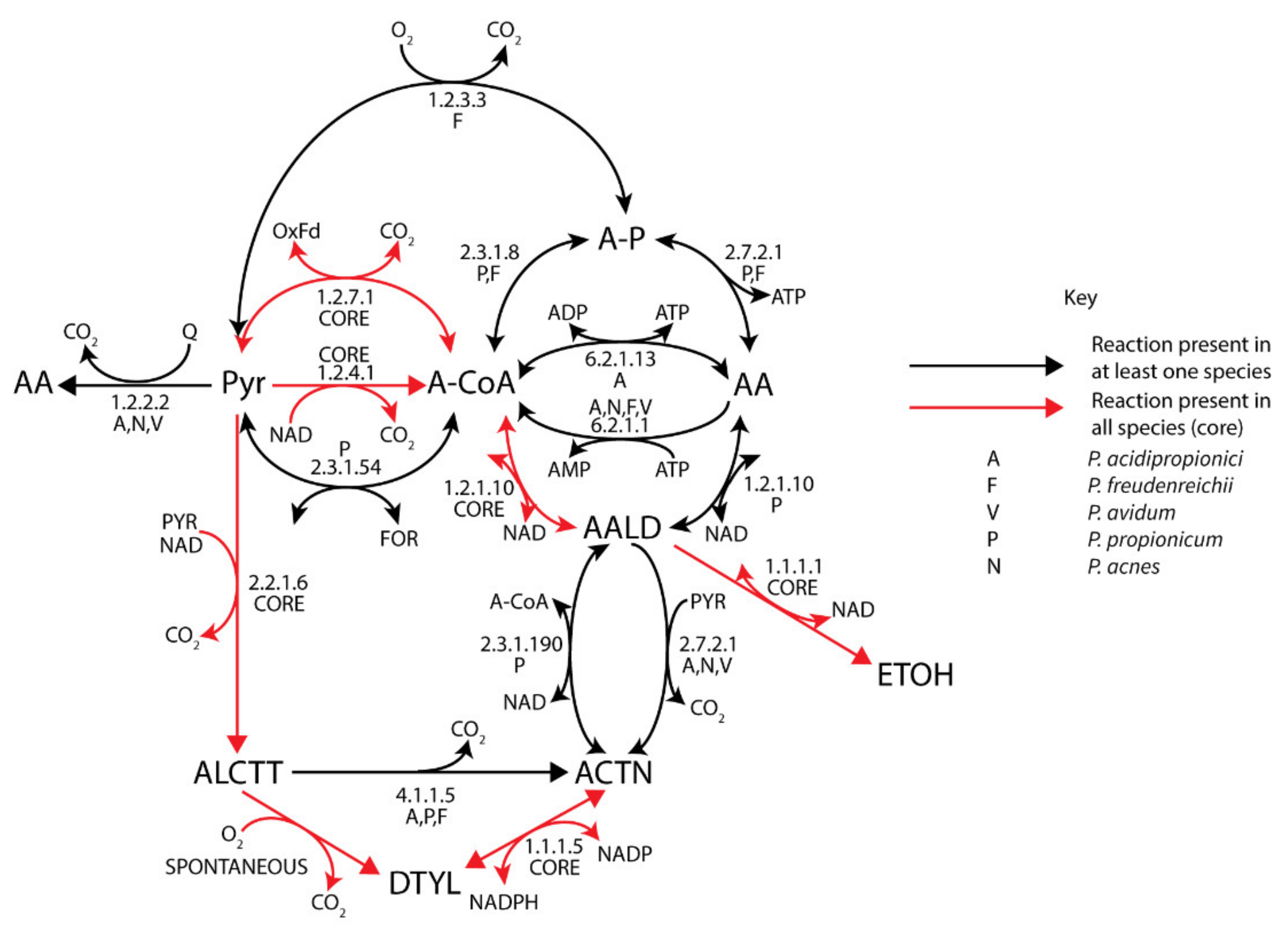
3.4.2. Acetate, Ethanol, and Acetoin Metabolism
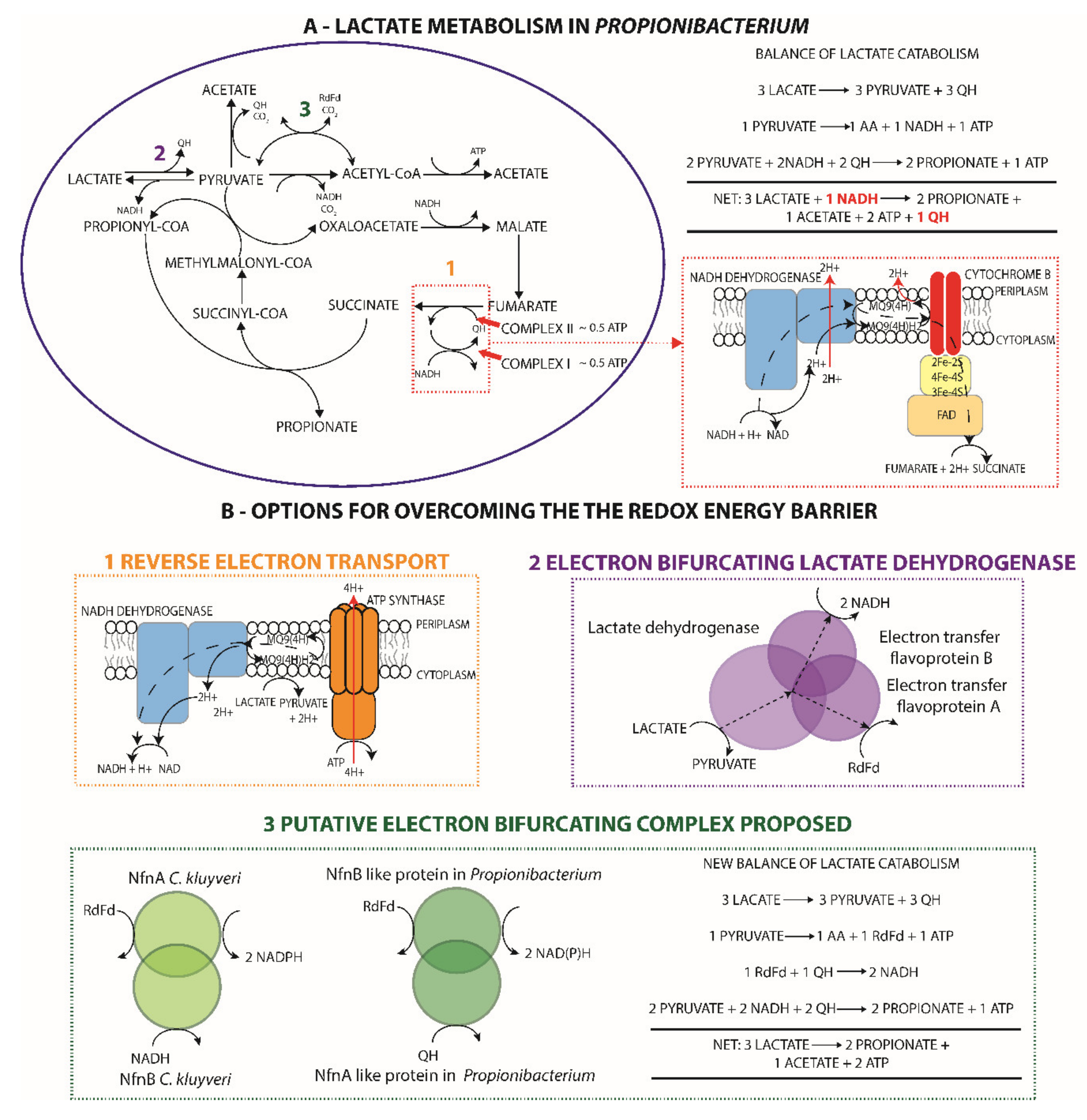
3.4.3. Lactate and Ferredoxin Metabolism
3.4.4. Pyruvate Metabolism
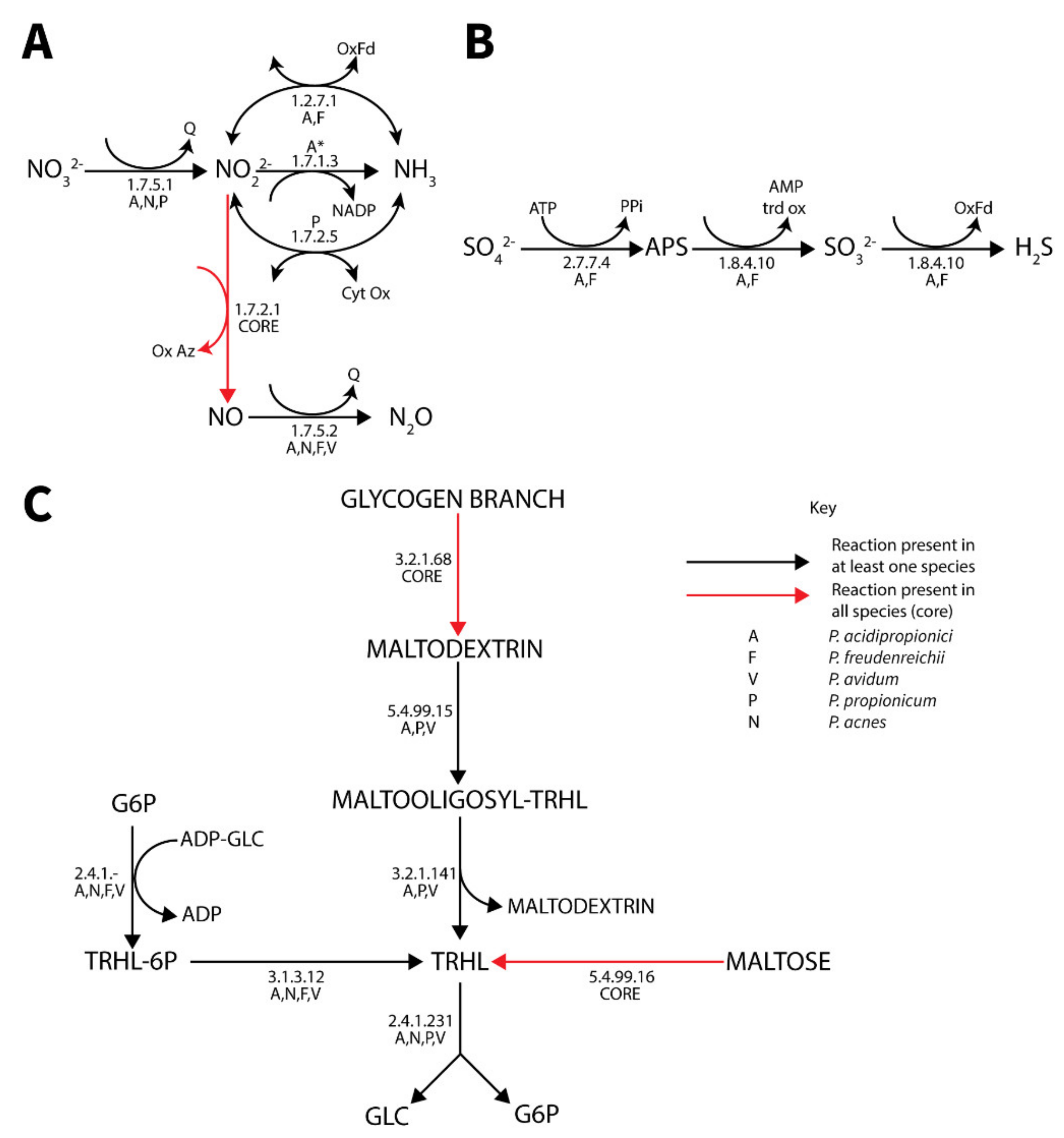
3.4.5. Respiration
3.4.6. Polyphosphate Metabolism
3.4.7. Trehalose Metabolism
3.4.8. Amino Acid Biosynthesis and Catabolism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seeliger, S.; Janssen, P.H.; Schink, B. Energetics and kinetics of lactate fermentation to acetate and propionate via methylmalonyl-CoA or acrylyl-CoA. FEMS Microbiol. Lett. 2002, 211, 65–70. [Google Scholar] [CrossRef]
- De Vries, W.; van Wyck-Kapteyn, W.M.C.; Stouthamer, A.H. Generation of ATP during Cytochrome-linked Anaerobic Electron Transport in Propionic Acid Bacteria. J. Gen. Microbiol. 1973, 76, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.F.P.; Kilian, M. The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus Propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, O.A.; Dekio, I.; Layton, A.M.; Li, H.; Hughes, H.; Morris, T.; Zouboulis, C.C.; Patrick, S. Why we continue to use the name Propionibacterium acnes. Br. J. Dermatol. 2018, 179, 1227. [Google Scholar] [CrossRef]
- Rabah, H.; Rosa do Carmo, F.; Jan, G. Dairy Propionibacteria: Versatile Probiotics. Microorganisms 2017, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Piwowarek, K.; Lipińska, E.; Hać-Szymańczuk, E.; Kieliszek, M.; Ścibisz, I. Propionibacterium spp.—source of propionic acid, vitamin B12, and other metabolites important for the industry. Appl. Microbiol. Biotechnol. 2018, 102, 515–538. [Google Scholar] [CrossRef]
- Turgay, M.; Bachmann, H.-P.; Irmler, S.; von Ah, U.; Fröhlich-Wyder, M.-T.; Falentin, H.; Deutsch, S.-M.; Jan, G.; Thierry, A. Propionibacterium spp. and Acidipropionibacterium spp. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780081005965. [Google Scholar]
- Rodriguez, B.A.; Stowers, C.C.; Pham, V.; Cox, B.M. The production of propionic acid, propanol and propylene via sugar fermentation: An industrial perspective on the progress, technical challenges and future outlook. Green Chem. 2014, 16, 1066–1076. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, R.; McCubbin, T.; Navone, L.; Stowers, C.; Nielsen, L.; Marcellin, E. Microbial Propionic Acid Production. Fermentation 2017, 3, 21. [Google Scholar] [CrossRef]
- Corvec, S. Clinical and Biological Features of Cutibacterium (Formerly Propionibacterium) avidum, an Underrecognized Microorganism. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef]
- Achermann, Y.; Goldstein, E.J.C.; Coenye, T.; Shirtliff, M.E. Propionibacterium acnes: From Commensal to Opportunistic Biofilm-Associated Implant Pathogen. Clin. Microbiol. Rev. 2014, 27, 419–440. [Google Scholar] [CrossRef]
- Siqueira, J.F. Periapical Actinomycosis and infection with Propionibacterium Propionicum. Endod. Top. 2003, 6, 78–95. [Google Scholar] [CrossRef]
- Chavali, A.K.; D’Auria, K.M.; Hewlett, E.L.; Pearson, R.D.; Papin, J.A. A metabolic network approach for the identification and prioritization of antimicrobial drug targets. Trends Microbiol. 2012, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, S.-M.; Le Bivic, P.; Hervé, C.; Madec, M.-N.; LaPointe, G.; Jan, G.; Le Loir, Y.; Falentin, H. Correlation of the capsular phenotype in Propionibacterium freudenreichii with the level of expression of gtf, a unique polysaccharide synthase-encoding gene. Appl. Environ. Microbiol. 2010, 76, 2740–2746. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, X.; Liu, L.; Shin, H.; Chen, R.R.; Li, J.; Du, G.; Chen, J. Development of a Propionibacterium-Escherichia coli shuttle vector for metabolic engineering of Propionibacterium jensenii, an efficient producer of propionic acid. Appl. Environ. Microbiol. 2013, 79, 4595–4602. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Cui, H.; Zhu, L.; Hu, Y.; Xu, X.; Li, S.; Huang, H. Enhanced propionic acid production from whey lactose with immobilized Propionibacterium acidipropionici and the role of trehalose synthesis in acid tolerance. Green Chem. 2015, 17, 250–259. [Google Scholar] [CrossRef]
- Frohnmeyer, E.; Deptula, P.; Nyman, T.A.; Laine, P.K.S.; Vihinen, H.; Paulin, L.; Auvinen, P.; Jokitalo, E.; Piironen, V.; Varmanen, P.; et al. Secretome profiling of Propionibacterium freudenreichii reveals highly variable responses even among the closely related strains. Microb. Biotechnol. 2018, 11, 510–526. [Google Scholar] [CrossRef]
- Yee, A.L.; Maillard, M.-B.; Roland, N.; Chuat, V.; Leclerc, A.; Pogačić, T.; Valence, F.; Thierry, A. Great interspecies and intraspecies diversity of dairy propionibacteria in the production of cheese aroma compounds. Int. J. Food Microbiol. 2014, 191, 60–68. [Google Scholar] [CrossRef]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef]
- Deptula, P.; Laine, P.K.; Roberts, R.J.; Smolander, O.-P.; Vihinen, H.; Piironen, V.; Paulin, L.; Jokitalo, E.; Savijoki, K.; Auvinen, P.; et al. De novo assembly of genomes from long sequence reads reveals uncharted territories of Propionibacterium freudenreichii. BMC Genom. 2017, 18, 790. [Google Scholar] [CrossRef]
- Angelova, M.; Kalajdziski, S.; Kocarev, L. Computational Methods for Gene Finding in Prokaryotes. In Proceedings of the Web Proceedings, Ohrid, Macedonia, 12–15 September 2010; pp. 11–20, ISSN 1857-7288. [Google Scholar]
- Suwannakham, S.; Huang, Y.; Yang, S.-T. Construction and characterization of ack knock-out mutants of Propionibacterium acidipropionici for enhanced propionic acid fermentation. Biotechnol. Bioeng. 2006, 94, 383–395. [Google Scholar] [CrossRef]
- Falentin, H.; Deutsch, S.-M.; Jan, G.; Loux, V.; Thierry, A.; Parayre, S.; Maillard, M.-B.; Dherbécourt, J.; Cousin, F.J.; Jardin, J.; et al. The complete genome of Propionibacterium freudenreichii CIRM-BIA1, a hardy actinobacterium with food and probiotic applications. PLoS ONE 2010, 5, e11748. [Google Scholar] [CrossRef] [PubMed]
- Parizzi, L.P.; Grassi, M.C.B.; Llerena, L.A.; Carazzolle, M.F.; Queiroz, V.L.; Lunardi, I.; Zeidler, A.F.; Teixeira, P.J.P.L.; Mieczkowski, P.; Rincones, J.; et al. The genome sequence of Propionibacterium acidipropionici provides insights into its biotechnological and industrial potential. BMC Genom. 2012, 13, 562. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2016, 35, 81–89. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Zhi, X.-Y.; Li, H.-W.; Klenk, H.-P.; Li, W.-J. Comparative Genomics of the Bacterial Genus Streptococcus Illuminates Evolutionary Implications of Species Groups. PLoS ONE 2014, 9, e101229. [Google Scholar] [CrossRef]
- Wassenaar, T.M.; Jun, S.-R.; Wanchai, V.; Patumcharoenpol, P.; Nookaew, I.; Schlum, K.; Leuze, M.R.; Ussery, D.W. Insights from Comparative Genomics of the Genus Salmonella. In Current Topics in Salmonella and Salmonellosis; InTech: London, UK, 2017. [Google Scholar]
- Steinway, S.N.; Biggs, M.B.; Loughran, T.P.; Papin, J.A.; Albert, R. Inference of Network Dynamics and Metabolic Interactions in the Gut Microbiome. PLoS Comput. Biol. 2015, 11, e1004338. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Arkin, A.P.; Stevens, R.L.; Cottingham, R.W.; Maslov, S.; Henry, C.S.; Dehal, P.; Ware, D.; Perez, F.; Harris, N.L.; Canon, S.; et al. The DOE Systems Biology Knowledgebase (KBase). bioRxiv 2016. [Google Scholar] [CrossRef]
- Luna-Flores, C.H.; Nielsen, L.K.; Marcellin, E. Genome Sequence of Propionibacterium acidipropionici ATCC 55737. Genome Announc. 2016, 4, e00248-16. [Google Scholar] [CrossRef] [PubMed]
- Ordogh, L.; Hunyadkurti, J.; Voros, A.; Horvath, B.; Szucs, A.; Urban, E.; Kereszt, A.; Kondorosi, E.; Nagy, I. Complete Genome Sequence of Propionibacterium avidum Strain 44067, Isolated from a Human Skin Abscess. Genome Announc. 2013, 1, e00337-13. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef]
- Brüggemann, H.; Henne, A.; Hoster, F.; Liesegang, H.; Wiezer, A.; Strittmatter, A.; Hujer, S.; Dürre, P.; Gottschalk, G. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science 2004, 305, 671–673. [Google Scholar] [CrossRef]
- Hunyadkürti, J.; Feltóti, Z.; Horváth, B.; Nagymihály, M.; Vörös, A.; McDowell, A.; Patrick, S.; Urbán, E.; Nagy, I. Complete genome sequence of Propionibacterium acnes type IB strain 6609. J. Bacteriol. 2011, 193, 4561–4562. [Google Scholar] [CrossRef]
- Vörös, A.; Horváth, B.; Hunyadkürti, J.; McDowell, A.; Barnard, E.; Patrick, S.; Nagy, I. Complete genome sequences of three Propionibacterium acnes isolates from the type IA(2) cluster. J. Bacteriol. 2012, 194, 1621–1622. [Google Scholar] [CrossRef]
- Horváth, B.; Hunyadkürti, J.; Vörös, A.; Fekete, C.; Urbán, E.; Kemény, L.; Nagy, I. Genome sequence of Propionibacterium acnes type II strain ATCC 11828. J. Bacteriol. 2012, 194, 202–203. [Google Scholar] [CrossRef]
- Minegishi, K.; Aikawa, C.; Furukawa, A.; Watanabe, T.; Nakano, T.; Ogura, Y.; Ohtsubo, Y.; Kurokawa, K.; Hayashi, T.; Maruyama, F.; et al. Complete Genome Sequence of a Propionibacterium acnes Isolate from a Sarcoidosis Patient. Genome Announc. 2013, 1. [Google Scholar] [CrossRef]
- Fitz-Gibbon, S.; Tomida, S.; Chiu, B.-H.; Nguyen, L.; Du, C.; Liu, M.; Elashoff, D.; Erfe, M.C.; Loncaric, A.; Kim, J.; et al. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Investig. Dermatol. 2013, 133, 2152–2160. [Google Scholar] [CrossRef]
- Brzuszkiewicz, E.; Weiner, J.; Wollherr, A.; Thürmer, A.; Hüpeden, J.; Lomholt, H.B.; Kilian, M.; Gottschalk, G.; Daniel, R.; Mollenkopf, H.-J.; et al. Comparative genomics and transcriptomics of Propionibacterium acnes. PLoS ONE 2011, 6, e21581. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Willenbrock, H.; Hallin, P.F.; Wassenaar, T.M.; Ussery, D.W. Characterization of probiotic Escherichia coli isolates with a novel pan-genome microarray. Genome Biol. 2007, 8, R267. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.J.L.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D. TreeView: An application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 1996, 12, 357–358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sutcliffe, I.C.; Shaw, N. An inositol containing lipomannan from Propionibacterium freudenreichii. FEMS Microbiol. Lett. 1989, 59, 249–251. [Google Scholar] [CrossRef]
- Cummins, C.S.; White, R.H. Isolation, identification, and synthesis of 2,3-diamino-2,3-dideoxyglucuronic acid: A component of Propionibacterium acnes cell wall polysaccharide. J. Bacteriol. 1983, 153, 1388–1393. [Google Scholar] [CrossRef]
- Moss, C.W.; Dowell, V.R.; Farshtchi, D.; Raines, L.J.; Cherry, W.B. Cultural characteristics and fatty acid composition of propionibacteria. J. Bacteriol. 1969, 97, 561–570. [Google Scholar] [CrossRef]
- Kim, M.; Sang Yi, J.; Kim, J.; Kim, J.-N.; Kim, M.W.; Kim, B.-G. Reconstruction of a high-quality metabolic model enables the identification of gene overexpression targets for enhanced antibiotic production in Streptomyces coelicolor A3(2). Biotechnol. J. 2014, 9, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Piveteau, P. Metabolism of lactate and sugars by dairy propionibacteria: A review. Lait 1999, 79, 23–41. [Google Scholar] [CrossRef]
- Scheer, M.; Grote, A.; Chang, A.; Schomburg, I.; Munaretto, C.; Rother, M.; Söhngen, C.; Stelzer, M.; Thiele, J.; Schomburg, D. BRENDA, the enzyme information system in 2011. Nucleic Acids Res. 2011, 39, D670–D676. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, E.; Mercer, T.R.; Licona-Cassani, C.; Palfreyman, R.W.; Dinger, M.E.; Steen, J.A.; Mattick, J.S.; Nielsen, L.K. Saccharopolyspora erythraea’s genome is organised in high-order transcriptional regions mediated by targeted degradation at the metabolic switch. BMC Genom. 2013, 14, 15. [Google Scholar] [CrossRef]
- Licona-Cassani, C.; Lim, S.; Marcellin, E.; Nielsen, L.K. Temporal Dynamics of the Saccharopolyspora erythraea Phosphoproteome. Mol. Cell. Proteom. 2014, 13, 1219–1230. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Etheridge, N.; Nouwens, A.S.; Dodd, P.R. SWATH analysis of the synaptic proteome in Alzheimer’s disease. Neurochem. Int. 2015, 87, 1–12. [Google Scholar] [CrossRef]
- Luna-Flores, C.H.; Palfreyman, R.W.; Krömer, J.O.; Nielsen, L.K.; Marcellin, E. Improved production of propionic acid using genome shuffling. Biotechnol. J. 2017, 12, 1600120. [Google Scholar] [CrossRef] [PubMed]
- Navone, L.; McCubbin, T.; Gonzalez-Garcia, R.A.; Nielsen, L.K.; Marcellin, E. Genome-scale model guided design of Propionibacterium for enhanced propionic acid production. Metab. Eng. Commun. 2018, 6, 1–12. [Google Scholar] [CrossRef]
- Kiatpapan, P.; Hashimoto, Y.; Nakamura, H.; Piao, Y.Z.; Ono, H.; Yamashita, M.; Murooka, Y. Characterization of pRGO1, a plasmid from Propionibacterium acidipropionici, and its use for development of a host-vector system in propionibacteria. Appl. Environ. Microbiol. 2000, 66, 4688–4695. [Google Scholar] [CrossRef]
- Wang, Z.; Ammar, E.M.; Zhang, A.; Wang, L.; Lin, M.; Yang, S.-T. Engineering Propionibacterium freudenreichii subsp. shermanii for enhanced propionic acid fermentation: Effects of overexpressing propionyl-CoA:Succinate CoA transferase. Metab. Eng. 2015, 27, 46–56. [Google Scholar] [CrossRef]
- Liu, L.; Zhuge, X.; Shin, H.; Chen, R.R.; Li, J.; Du, G.; Chen, J. Improved Production of Propionic Acid in Propionibacterium jensenii via Combinational Overexpression of Glycerol Dehydrogenase and Malate Dehydrogenase from Klebsiella pneumoniae. Appl. Environ. Microbiol. 2015, 81, 2256–2264. [Google Scholar] [CrossRef]
- Luna-Flores, C.H.; Stowers, C.C.; Cox, B.M.; Nielsen, L.K.; Marcellin, E. Linking genotype and phenotype in an economically viable propionic acid biosynthesis process. Biotechnol. Biofuels 2018, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Stowers, C.C.; Cox, B.M.; Rodriguez, B. A Development of an industrializable fermentation process for propionic acid production. J. Ind. Microbiol. Biotechnol. 2014, 41, 837–852. [Google Scholar] [CrossRef]
- Bott, M.; Pfister, K.; Burda, P.; Kalbermatter, O.; Woehlke, G.; Dimroth, P. Methylmalonyl-CoA Decarboxylase from Propionigenium Modestum. Cloning and Sequencing of the Structural Genes and Purification of the Enzyme Complex. Eur. J. Biochem. 1997, 250, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, F.; Xu, H.; Wu, B.; Li, H.; Li, S.; Ouyang, P. Green and economical production of propionic acid by Propionibacterium freudenreichii CCTCC M207015 in plant fibrous-bed bioreactor. Bioresour. Technol. 2011, 102, 6141–6146. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, L.; Hrůzová, K.; Rova, U.; Christakopoulos, P. Biological Production of 3-Hydroxypropionic Acid: An Update on the Current Status. Fermentation 2018, 4, 13. [Google Scholar] [CrossRef]
- Toraya, T.; Kuno, S.; Fukui, S. Distribution of coenzyme B12-dependent diol dehydratase and glycerol dehydratase in selected genera of Enterobacteriaceae and Propionibacteriaceae. J. Bacteriol. 1980, 141, 1439–1442. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, K.; Hojo, K.; Katakura, Y.; Ninomiya, K.; Shioya, S. Aerobic culture of Propionibacterium freudenreichii ET-3 can increase production ratio of 1,4-dihydroxy-2-naphthoic acid to menaquinone. J. Biosci. Bioeng. 2006, 101, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Fan, C.; Sinha, S.; Bobik, T.A. The PduQ Enzyme Is an Alcohol Dehydrogenase Used to Recycle NAD+ Internally within the Pdu Microcompartment of Salmonella enterica. PLoS ONE 2012, 7, e47144. [Google Scholar] [CrossRef]
- Lee, I.H.; Fredrickson, A.G.; Tsuchiya, H.M. Diauxic Growth of Propionibacterium shermanii. Appl. Microbiol. 1974, 28, 831–835. [Google Scholar]
- Bott, M.; Niebisch, A. The respiratory chain of Corynebacterium glutamicum. J. Biotechnol. 2003, 104, 129–153. [Google Scholar] [CrossRef]
- Chai, Y.; Kolter, R.; Losick, R. A Widely Conserved Gene Cluster Required for Lactate Utilization in Bacillus subtilis and Its Involvement in Biofilm Formation. J. Bacteriol. 2009, 191, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, G.E.; Rodionov, D.A.; Yang, C.; Li, X.; Osterman, A.L.; Dervyn, E.; Geydebrekht, O.V.; Reed, S.B.; Romine, M.F.; Collart, F.R.; et al. Genomic reconstruction of Shewanella oneidensis MR-1 metabolism reveals a previously uncharacterized machinery for lactate utilization. Proc. Natl. Acad. Sci. USA 2009, 106, 2874–2879. [Google Scholar] [CrossRef]
- Sone, N. The redox reactions in propionic acid fermentation. 3. Enzymatic properties of NAD-independent glycerol-phosphate dehydrogenase from Propionibacterium arabinosum. J. Biochem. 1973, 74, 297–305. [Google Scholar]
- Schwartz, A.C.; Sporkenbach, J. The electron transport system of the anaerobic Propionibacterium shermanii. Arch. Microbiol. 1975, 102, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Crow, V.L. Utilization of Lactate Isomers by Propionibacterium freudenreichii subsp. shermanii: Regulatory Role for Intracellular Pyruvate. Appl. Environ. Microbiol. 1986, 52, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Elbehti, A.; Brasseur, G.; Lemesle-Meunier, D. First evidence for existence of an uphill electron transfer through the bc(1) and NADH-Q oxidoreductase complexes of the acidophilic obligate chemolithotrophic ferrous ion-oxidizing bacterium Thiobacillus ferrooxidans. J. Bacteriol. 2000, 182, 3602–3606. [Google Scholar] [CrossRef]
- Lücker, S.; Nowka, B.; Rattei, T.; Spieck, E.; Daims, H. The Genome of Nitrospina gracilis Illuminates the Metabolism and Evolution of the Major Marine Nitrite Oxidizer. Front. Microbiol. 2013, 4, 27. [Google Scholar] [CrossRef]
- Weghoff, M.C.; Bertsch, J.; Müller, V. A novel mode of lactate metabolism in strictly anaerobic bacteria. Environ. Microbiol. 2015, 17, 670–677. [Google Scholar] [CrossRef]
- Ledbetter, R.N.; Garcia Costas, A.M.; Lubner, C.E.; Mulder, D.W.; Tokmina-Lukaszewska, M.; Artz, J.H.; Patterson, A.; Magnuson, T.S.; Jay, Z.J.; Duan, H.D.; et al. The Electron Bifurcating FixABCX Protein Complex from Azotobacter vinelandii: Generation of Low-Potential Reducing Equivalents for Nitrogenase Catalysis. Biochemistry 2017, 56, 4177–4190. [Google Scholar] [CrossRef]
- Buckel, W.; Thauer, R.K. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim. Biophys. Acta 2013, 1827, 94–113. [Google Scholar] [CrossRef]
- Chiba, Y.; Kamikawa, R.; Nakada-Tsukui, K.; Saito-Nakano, Y.; Nozaki, T. Discovery of PPi-type Phosphoenolpyruvate Carboxykinase Genes in Eukaryotes and Bacteria. J. Biol. Chem. 2015, 290, 23960–23970. [Google Scholar] [CrossRef]
- Zhang, H.; Ishige, K.; Kornberg, A. A polyphosphate kinase (PPK2) widely conserved in bacteria. Proc. Natl. Acad. Sci. USA 2002, 99, 16678–16683. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; Tang, C.M. The role of the exopolyphosphatase PPX in avoidance by Neisseria meningitidis of complement-mediated killing. J. Biol. Chem. 2010, 285, 34259–34268. [Google Scholar] [CrossRef]
- Cardoso, F.S.; Castro, R.F.; Borges, N.; Santos, H. Biochemical and genetic characterization of the pathways for trehalose metabolism in Propionibacterium freudenreichii, and their role in stress response. Microbiology 2007, 153, 270–280. [Google Scholar] [CrossRef]
- Thierry, A.; Maillard, M.-B.; Yvon, M. Conversion of L-leucine to isovaleric acid by Propionibacterium freudenreichii TL 34 and ITGP23. Appl. Environ. Microbiol. 2002, 68, 608–615. [Google Scholar] [CrossRef]
- Carlier, J.P.; Sellier, N. Gas chromatographic-mass spectral studies after methylation of metabolites produced by some anaerobic bacteria in spent media. J. Chromatogr. 1989, 493, 257–273. [Google Scholar] [CrossRef]
- Deptula, P.; Smolander, O.-P.; Laine, P.; Roberts, R.J.; Edelmann, M.; Peltola, P.; Piironen, V.; Paulin, L.; Storgårds, E.; Savijoki, K.; et al. Acidipropionibacterium virtanenii sp. nov., isolated from malted barley. Int. J. Syst. Evol. Microbiol. 2018, 68, 3175–3183. [Google Scholar] [CrossRef]
- Bernier, A.-M.; Bernard, K. Whole-Genome Sequences of Propionibacterium australiense NML (LCDC) 98A072T and NML (LCDC) 98A078, Associated with Granulomatous Bovine Lesions. Microbiol. Resour. Announc. 2018, 7. [Google Scholar] [CrossRef]
- Dekio, I.; Sakamoto, M.; Suzuki, T.; Yuki, M.; Kinoshita, S.; Murakami, Y.; Ohkuma, M. Cutibacterium modestum sp. nov., isolated from meibum of human meibomian glands, and emended descriptions of Cutibacterium granulosum and Cutibacterium namnetense. Int. J. Syst. Evol. Microbiol. 2020, 70, 2457–2462. [Google Scholar] [CrossRef]
- Gaucher, F.; Kponouglo, K.; Rabah, H.; Bonnassie, S.; Ossemond, J.; Pottier, S.; Jardin, J.; Briard-Bion, V.; Marchand, P.; Blanc, P.; et al. Propionibacterium freudenreichii CIRM-BIA 129 Osmoadaptation Coupled to Acid-Adaptation Increases Its Viability During Freeze-Drying. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Cotter, P.D.; Hill, C. Surviving the Acid Test: Responses of Gram-Positive Bacteria to Low pH. Microbiol. Mol. Biol. Rev. 2003, 67, 429–453. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Accession | Reference | Metabolic Reconstruction |
|---|---|---|---|---|
| Dairy propionibacteria (Acidipropionibacterium, Propioinibacterium) | ||||
| A. Acidipropionici | ATCC 4875 | NC_019395.1 | [24] | Yes |
| ATCC 55737 | NZ_CP014352.1 | [35] | Yes | |
| P. freudenreichii | CIRM-BIA1 | NZ_CP010341.1 | [23] | Yes |
| Commensal propionibacteria (Cutibacterium, Pseudopropionibacterium) | ||||
| C. avidum | 44067 | NC_021064.1 | [36] | Yes |
| P. propionicum | F0230a | NC_018142.1 | [37,38] | Yes |
| C. acnes | KPA171202 | NC_006085.1 | [39] | No |
| 6609 | NC_017535.1 | [40] | Yes | |
| TypeIA2 P.acn17 | NC_016512.1 | [41] | No | |
| TypeIA2 P.acn31 | NC_016511.1 | [41] | No | |
| TypeIA2 P.acn33 | NC_016516.1 | [41] | No | |
| ATCC 11828 | NC_017550.1 | [42] | No | |
| C1 | NC_018707.1 | [43] | No | |
| HL096PA1 | NC_021085.1 | [44] | No | |
| SK137 | NC_014039.1 | [37,38] | No | |
| 266 | NC_017534.1 | [45] | No | |
| hdn-1 | NZ_CP006032.1 | (Nagy et al., unpublished) | No | |
| Plasmids | Reference | |
|---|---|---|
| pRGO1 | Plasmid from P. acidipropionici | Kiatpapan [62] |
| pBRPprp_gfpuv | PBR322-based plasmid. gfpUV expression under the control of prpR promoter. AmpR | Lab collection |
| pPAC_Cas9 | pRGO1-derived plasmid harboring optimized Cas9. EryR. ApraR | This work |
| pCas9_nifJ | pPAC_Ca9-derived plasmid. Contains gRNA region for knockout of nifJ | This work |
| Primers | ||
| ackUP-HA_fwd | tttttaagcttcccgTCTCGCCGCTACCGCGCTTG | |
| ackUP-HA_rev | ggatagcgtcgccgtACGCCGCTGGCCGGCCTG | |
| ack-gfpUV_fwd | gccggccagcggcgtACGGCGACGCTATCCCCA | |
| ack-gfpUV_rev | tcggcgagctcaaccTTATTATTTGTAGAGCTCATCCATGCCATG | |
| ack-DW-HA_fwd | ctctacaaataataaGGTTGAGCTCGCCGAGGTCG | |
| ack-DW-HA_rev | aattggagctccaccgcggtggcggccgctCGGAGAACCCGGTGGCCG | |
| ack conf_fwd | CCGAGCATTCCCGAGTTC | |
| ack conf_rev | CTTCGACACCGCCTTCTTC | |
| 20 nt region | ccgccgggcgcaccaacctgTGG (PAM region shown in bold not included in gRNA scaffold) | |
| Model | P. acidi. 4875 | P. acidi. 55737 | P. freud | P. avidum | P. prop | P. acnes |
|---|---|---|---|---|---|---|
| Reactions added | 250 | 239 | 219 | 237 | 213 | 226 |
| Transporters added | 117 | 121 | 121 | 131 | 131 | 127 |
| Reactions removed | 26 | 22 | 29 | 21 | 22 | 19 |
| GPR altered | 139 | 141 | 107 | 119 | 111 | 122 |
| Total reactions | 1050 | 1056 | 933 | 962 | 977 | 952 |
| Total transporters | 242 | 241 | 217 | 229 | 218 | 231 |
| Reactions without GPR | 55 | 45 | 49 | 56 | 49 | 45 |
| Transporters without GPR | 103 | 103 | 109 | 112 | 115 | 109 |
| Database Modifications | |
|---|---|
| Reaction directionality changes | 116 |
| New reactions added to database | 105 |
| New metabolites added to database | 28 |
| Nutrient Requirement | P. acidipropionici | P. freudenreichii | P. avidum | P. propionicum | P. acnes | |
|---|---|---|---|---|---|---|
| Vitamins | Biotin | Biotin | Biotin | Pimelate | Biotin | Pimelate |
| Pantothenate | Pantothenate | Pantothenate | Pantothenate | Pantothenate | Pantothenate | |
| NAD | Nicotinate | Nicotinate | ||||
| Thiamin | Thiamin | Thiamin | Thiamin | |||
| Riboflavin | Riboflavin | |||||
| Pyridoxal | Pyridoxal | |||||
| Folate | p-aminobenzoate | |||||
| Amino acids | Methionine | Methionine | ||||
| Isoleucine | Isoleucine | |||||
| Leucine | Valine | |||||
| Valine | Valine | |||||
| Tryptophan | Tryptophan | |||||
| Phenylalanine | Phenylalanine | |||||
| Others | CTP | CMP | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCubbin, T.; Gonzalez-Garcia, R.A.; Palfreyman, R.W.; Stowers, C.; Nielsen, L.K.; Marcellin, E. A Pan-Genome Guided Metabolic Network Reconstruction of Five Propionibacterium Species Reveals Extensive Metabolic Diversity. Genes 2020, 11, 1115. https://doi.org/10.3390/genes11101115
McCubbin T, Gonzalez-Garcia RA, Palfreyman RW, Stowers C, Nielsen LK, Marcellin E. A Pan-Genome Guided Metabolic Network Reconstruction of Five Propionibacterium Species Reveals Extensive Metabolic Diversity. Genes. 2020; 11(10):1115. https://doi.org/10.3390/genes11101115
Chicago/Turabian StyleMcCubbin, Tim, R. Axayacatl Gonzalez-Garcia, Robin W. Palfreyman, Chris Stowers, Lars K. Nielsen, and Esteban Marcellin. 2020. "A Pan-Genome Guided Metabolic Network Reconstruction of Five Propionibacterium Species Reveals Extensive Metabolic Diversity" Genes 11, no. 10: 1115. https://doi.org/10.3390/genes11101115
APA StyleMcCubbin, T., Gonzalez-Garcia, R. A., Palfreyman, R. W., Stowers, C., Nielsen, L. K., & Marcellin, E. (2020). A Pan-Genome Guided Metabolic Network Reconstruction of Five Propionibacterium Species Reveals Extensive Metabolic Diversity. Genes, 11(10), 1115. https://doi.org/10.3390/genes11101115