Molecular Alterations in Thyroid Cancer: From Bench to Clinical Practice

 , , , ,
, , , ,
Abstract
1. Introduction
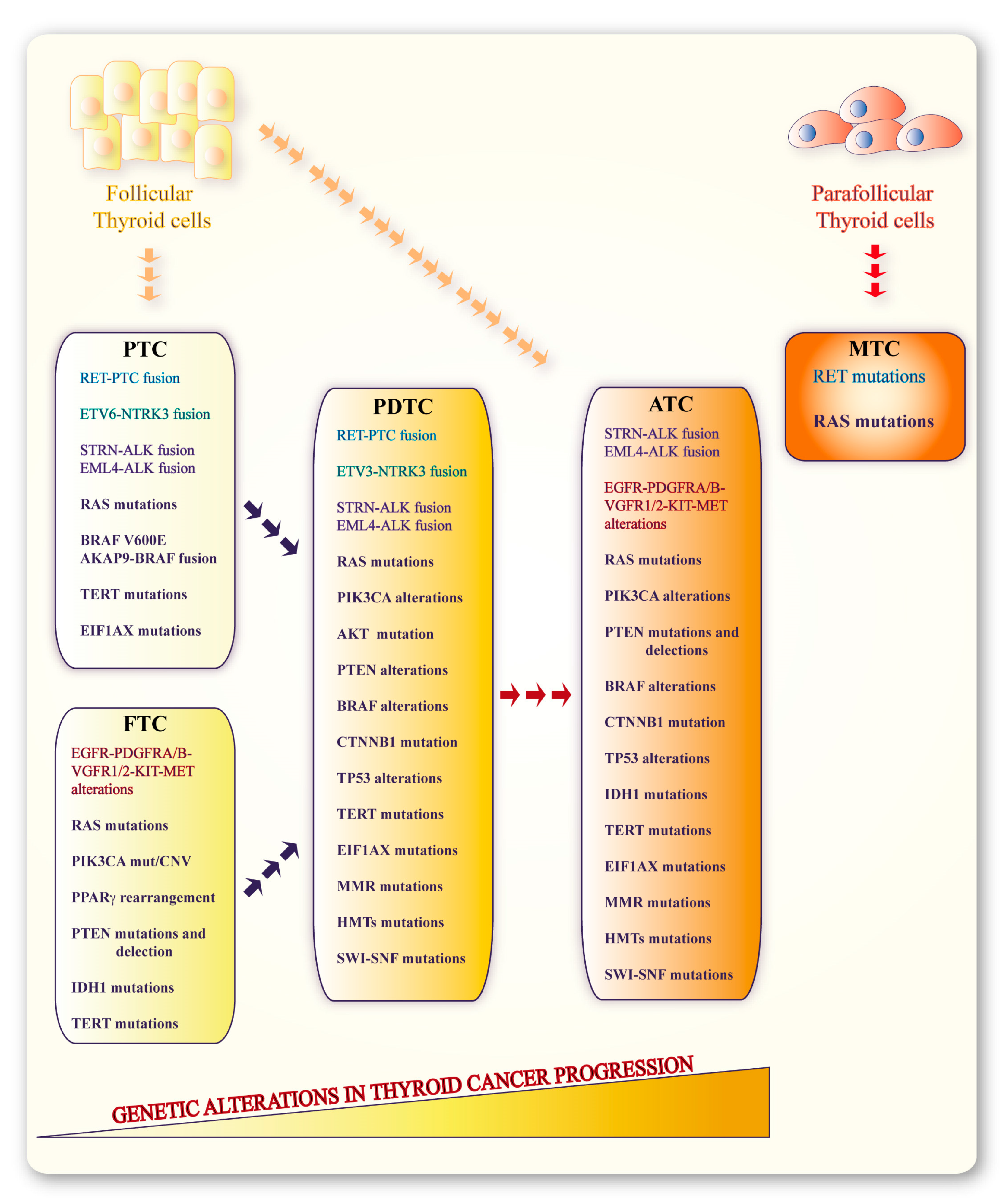
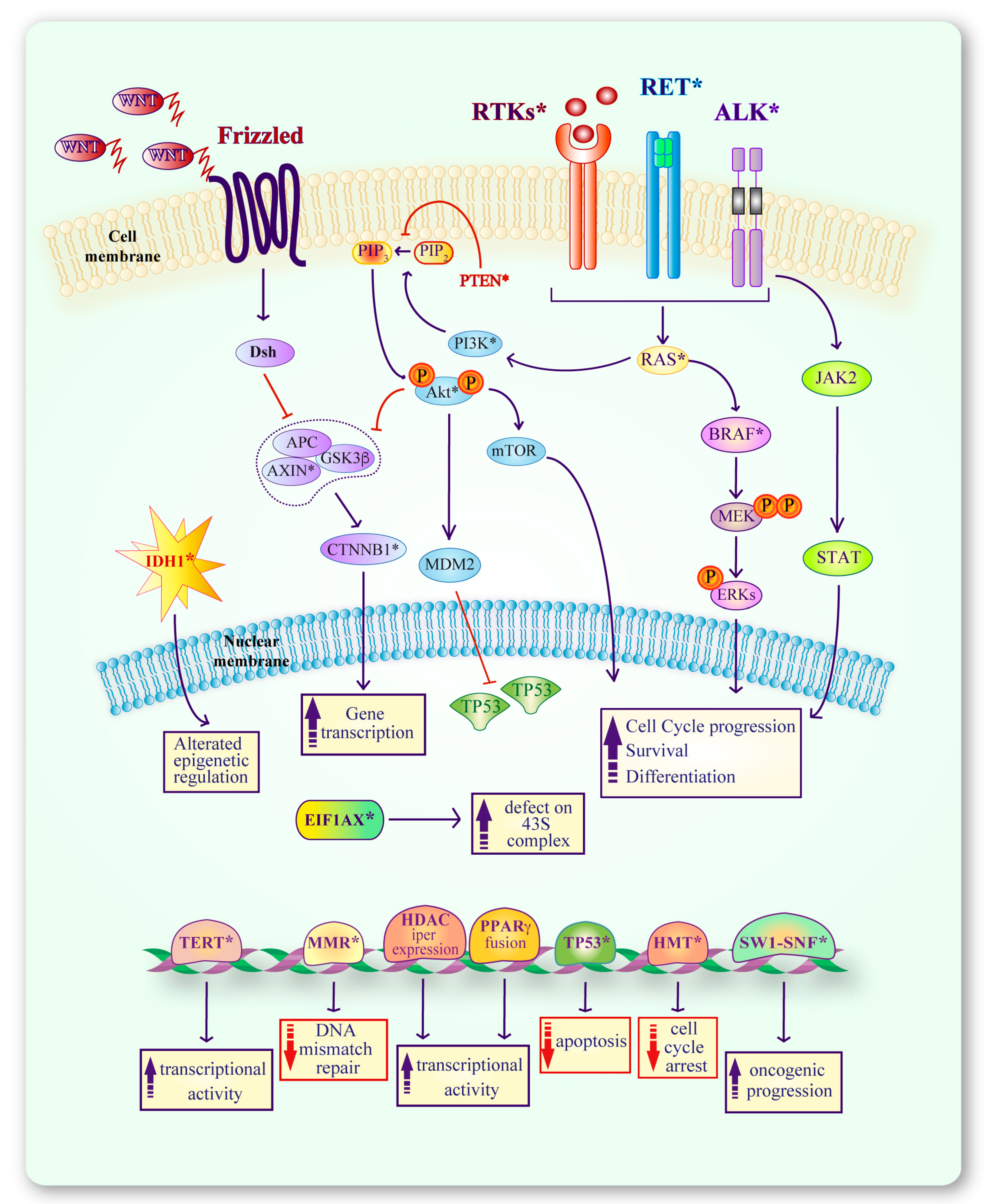
2. Molecular Alterations in Thyroid Cancers
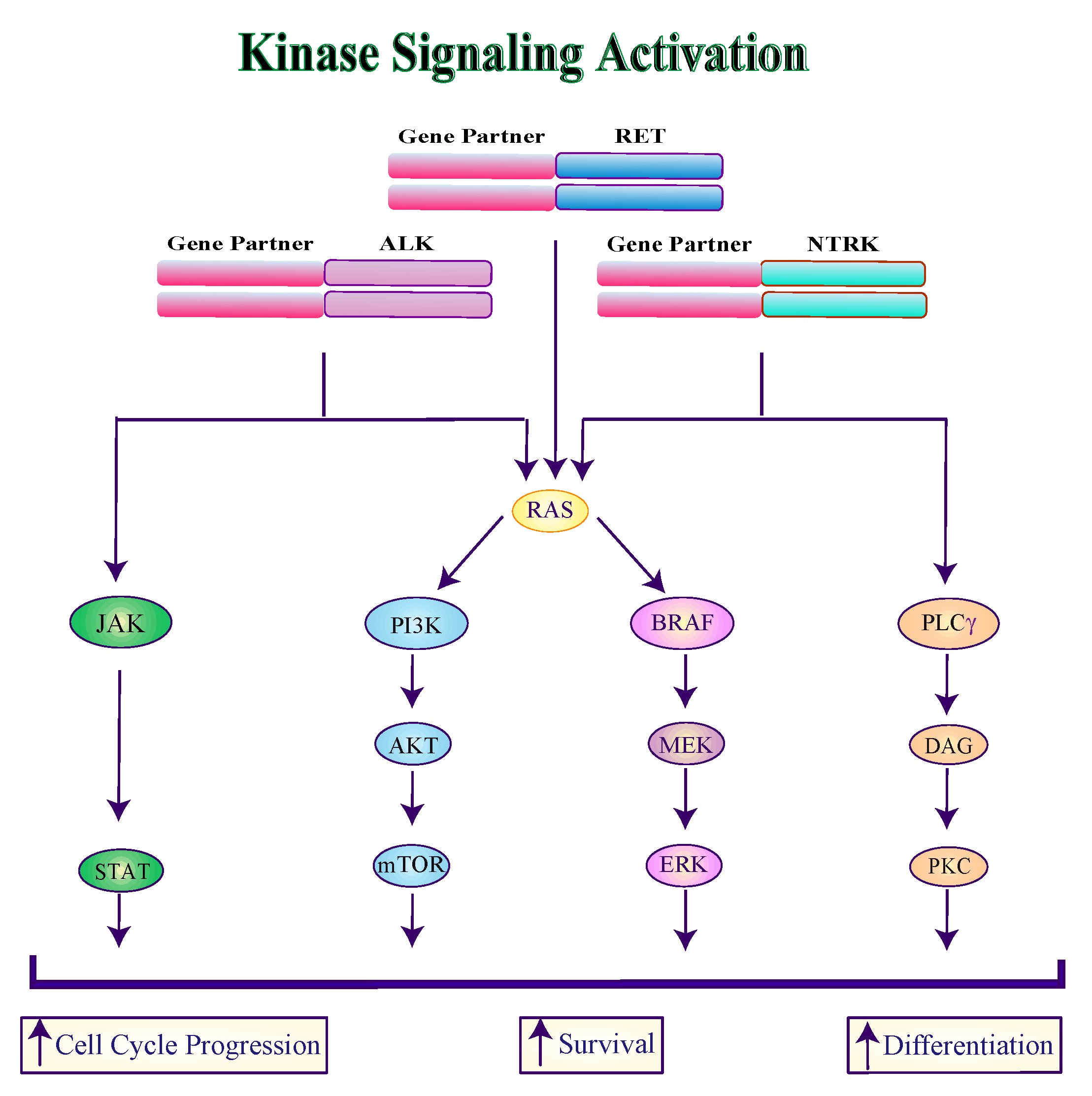
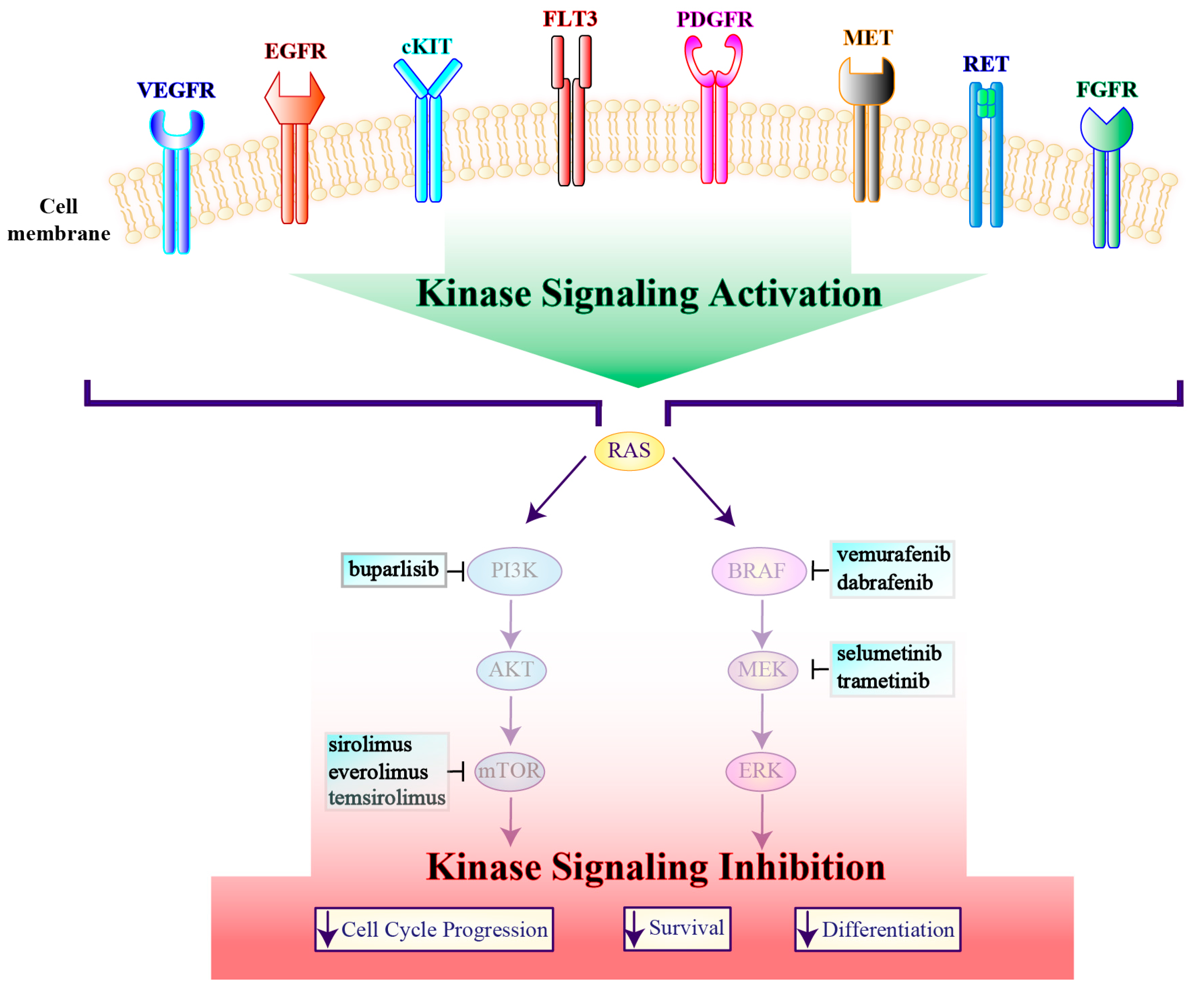
2.1. Alterations in RTKs
2.2. Alterations in the PI3K Pathway
2.3. Alterations in the MAPK Pathway
2.4. Alterations in the WNT Pathway
2.5. Alterations in the TP53 Pathway
2.6. Other Molecular Alterations in Thyroid Cancer
3. Targeted Therapies in Thyroid Cancer
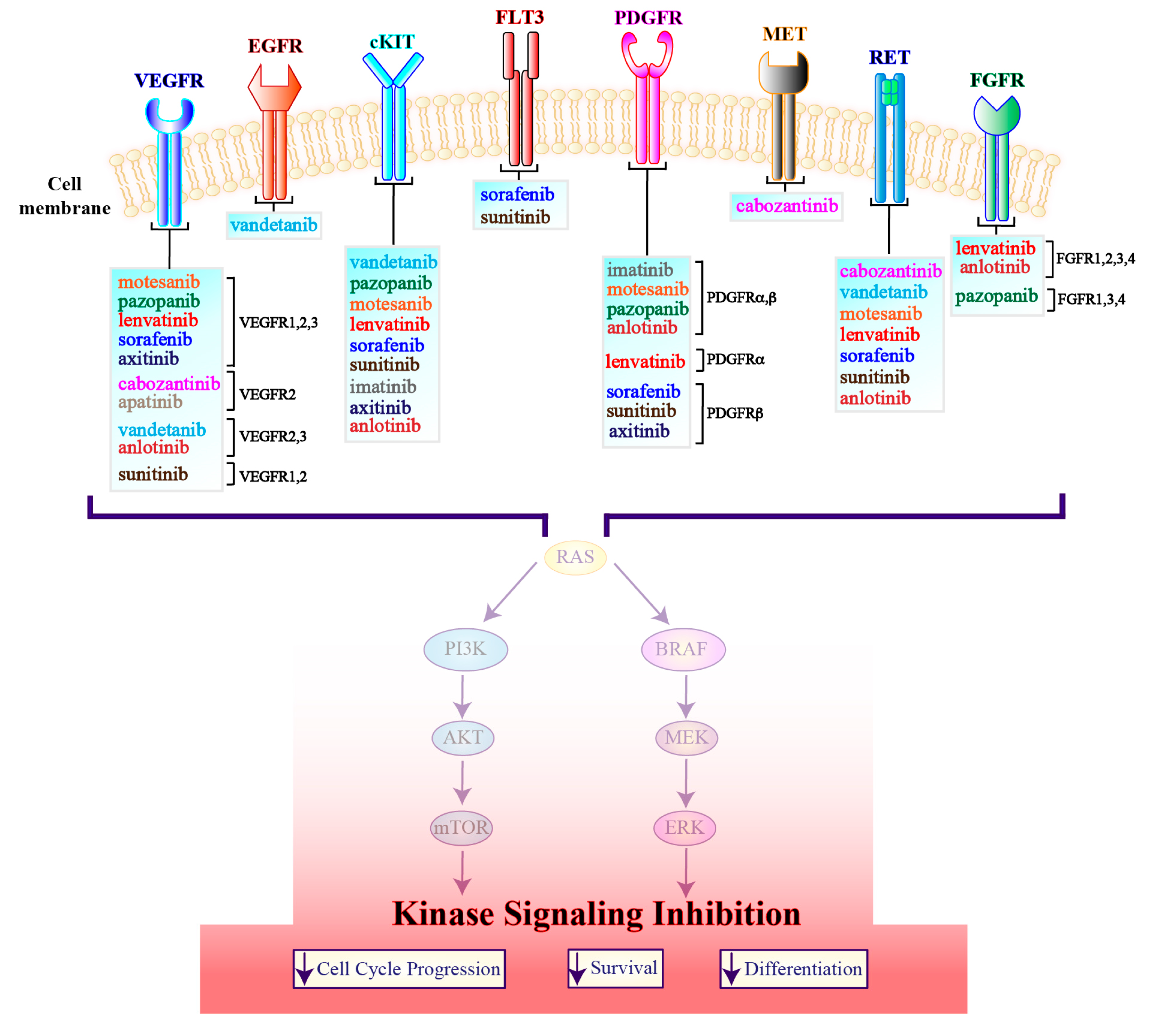
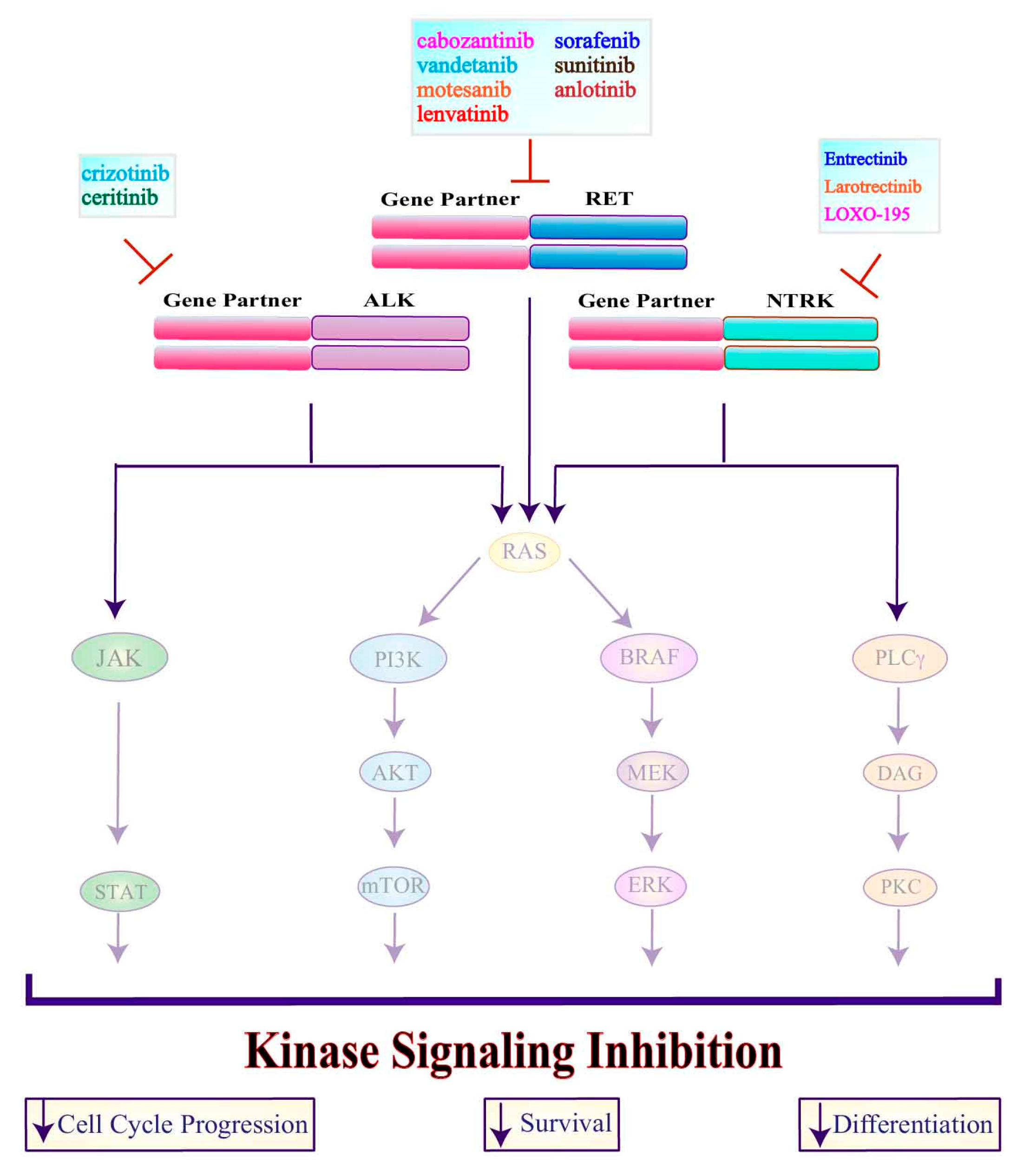
3.1. Tyrosine Kinase Inhibitors
3.1.1. Multi-Target Agents
3.1.2. Single-Target Agents
3.1.3. ALK Inhibitors
3.1.4. NTRK Inhibitors
3.2. PI3K/AKT/mTOR Pathway Inhibitors
3.3. PPAR-γ Agonist—Efatutazone
3.4. Histone Deacetylase Inhibitors—Valproic Acid
4. Immunotherapy Landscape in Thyroid Cancer
Immune Checkpoint Inhibitors
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- US Preventive Services Task Force; Bibbins-Domingo, K.; Grossman, D.C.; Curry, S.J.; Barry, M.J.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; Krist, A.H.; et al. Screening for Thyroid Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2017, 317, 1882–1887. [Google Scholar] [CrossRef] [PubMed]
- Pellegriti, G.; Frasca, F.; Regalbuto, C.; Squatrito, S.; Vigneri, R. Worldwide increasing incidence of thyroid cancer: Update on epidemiology and risk factors. J. Cancer Epidemiol. 2013, 2013, 965212. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, R.; Malandrino, P.; Giani, F.; Russo, M.; Vigneri, P. Heavy metals in the volcanic environment and thyroid cancer. Mol. Cell. Endocrinol. 2017, 457, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, R.; Malandrino, P.; Vigneri, P. The changing epidemiology of thyroid cancer: Why is incidence increasing? Curr. Opin. Oncol. 2015, 27, 7. [Google Scholar] [CrossRef] [PubMed]
- Peacock, M. Calcium metabolism in health and disease. Clin. J. Am. Soc. Nephrol. 2010, 5 (Suppl. S1), S23–S30. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- De la Fouchardiere, C.; Decaussin-Petrucci, M.; Berthiller, J.; Descotes, F.; Lopez, J.; Lifante, J.C.; Peix, J.L.; Giraudet, A.L.; Delahaye, A.; Masson, S.; et al. Predictive factors of outcome in poorly differentiated thyroid carcinomas. Eur. J. Cancer 2018, 92, 40–47. [Google Scholar] [CrossRef]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef]
- Randle, R.W.; Balentine, C.J.; Leverson, G.E.; Havlena, J.A.; Sippel, R.S.; Schneider, D.F.; Pitt, S.C. Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery 2017, 161, 137–146. [Google Scholar] [CrossRef]
- Schneider, D.F.; Chen, H. New developments in the diagnosis and treatment of thyroid cancer. CA Cancer J. Clin. 2013, 63, 374–394. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.I. Thyroid carcinoma. Lancet 2003, 361, 501–511. [Google Scholar] [CrossRef]
- Sampson, E.; Brierley, J.D.; Le, L.W.; Rotstein, L.; Tsang, R.W. Clinical management and outcome of papillary and follicular (differentiated) thyroid cancer presenting with distant metastasis at diagnosis. Cancer 2007, 110, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: Benefits and limits of radioiodine therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Brose, M.; Elisei, R.; Leboulleux, S.; Luster, M.; Pitoia, F.; Pacini, F. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014, 2, 356–358. [Google Scholar] [CrossRef]
- Faugeras, L.; Pirson, A.S.; Donckier, J.; Michel, L.; Lemaire, J.; Vandervorst, S.; D’Hondt, L. Refractory thyroid carcinoma: Which systemic treatment to use? Ther. Adv. Med. Oncol. 2018, 10, 1758834017752853. [Google Scholar] [CrossRef] [PubMed]
- Tumino, D.; Frasca, F.; Newbold, K. Updates on the Management of Advanced, Metastatic, and Radioiodine Refractory Differentiated Thyroid Cancer. Front. Endocrinol. (Lausanne) 2017, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Priya, S.R.; Dravid, C.S.; Digumarti, R.; Dandekar, M. Targeted Therapy for Medullary Thyroid Cancer: A Review. Front. Oncol. 2017, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, G.; Visani, M.; Repaci, A.; Rhoden, K.J.; de Biase, D.; Pession, A.; Giovanni, T. Molecular pathology of thyroid tumours of follicular cells: A review of genetic alterations and their clinicopathological relevance. Histopathology 2018, 72, 6–31. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, J.; Gawlik, T.; Jarzab, B. Advances in small molecule therapy for treating metastatic thyroid cancer. Expert Opin. Pharmacother. 2017, 18, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.N.; Zafereo, M.; Dadu, R.; Busaidy, N.L.; Hess, K.; Cote, G.J.; Williams, M.D.; William, W.N.; Sandulache, V.; Gross, N.; et al. Patterns of Treatment Failure in Anaplastic Thyroid Carcinoma. Thyroid 2017, 27, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Tirro, E.; Stella, S.; Frasca, F.; Vella, V.; Sciacca, L.; Pennisi, M.S.; Vitale, S.R.; Puma, A.; Romano, C.; et al. Effect of Combined Epigenetic Treatments and Ectopic NIS Expression on Undifferentiated Thyroid Cancer Cells. Anticancer Res. 2018, 38, 6653–6662. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Vigneri, P.; Fallica, M.; Fidilio, A.; Aloisi, A.; Frasca, F.; Manzella, L. IRF5 promotes the proliferation of human thyroid cancer cells. Mol. Cancer 2012, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R. The Emerging Role of Insulin Receptor Isoforms in Thyroid Cancer: Clinical Implications and New Perspectives. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Puppin, C.; Damante, G.; Vigneri, R.; Sanfilippo, M.; Vigneri, P.; Tell, G.; Frasca, F. DeltaNp73alpha inhibits PTEN expression in thyroid cancer cells. Int. J. Cancer 2009, 124, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Schagdarsurengin, U.; Gimm, O.; Dralle, H.; Hoang-Vu, C.; Dammann, R. CpG island methylation of tumor-related promoters occurs preferentially in undifferentiated carcinoma. Thyroid 2006, 16, 633–642. [Google Scholar] [CrossRef]
- Zarkesh, M.; Zadeh-Vakili, A.; Azizi, F.; Foroughi, F.; Akhavan, M.M.; Hedayati, M. Altered Epigenetic Mechanisms in Thyroid Cancer Subtypes. Mol. Diagn. Ther. 2018, 22, 41–56. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef] [PubMed]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef] [PubMed]
- Manzella, L.; Massimino, M.; Stella, S.; Tirro, E.; Pennisi, M.S.; Martorana, F.; Motta, G.; Vitale, S.R.; Puma, A.; Romano, C.; et al. Activation of the IGF Axis in Thyroid Cancer: Implications for Tumorigenesis and Treatment. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Yakushina, V.D.; Lerner, L.V.; Lavrov, A.V. Gene Fusions in Thyroid Cancer. Thyroid 2018, 28, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Demeure, M.J.; Aziz, M.; Rosenberg, R.; Gurley, S.D.; Bussey, K.J.; Carpten, J.D. Whole-genome sequencing of an aggressive BRAF wild-type papillary thyroid cancer identified EML4-ALK translocation as a therapeutic target. World J. Surg. 2014, 38, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.G.; Dias-Santagata, D.; Sadow, P.M.; Lynch, K.D.; Lubitz, C.; Donovan, S.E.; Zheng, Z.; Le, L.; Iafrate, A.J.; Daniels, G.H. Identification of oncogenic mutations and gene fusions in the follicular variant of papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E2457–E2462. [Google Scholar] [CrossRef]
- Kelly, L.M.; Barila, G.; Liu, P.; Evdokimova, V.N.; Trivedi, S.; Panebianco, F.; Gandhi, M.; Carty, S.E.; Hodak, S.P.; Luo, J.; et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4233–4238. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Murugan, A.K.; Xing, M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011, 71, 4403–4411. [Google Scholar] [CrossRef]
- Perot, G.; Soubeyran, I.; Ribeiro, A.; Bonhomme, B.; Savagner, F.; Boutet-Bouzamondo, N.; Hostein, I.; Bonichon, F.; Godbert, Y.; Chibon, F. Identification of a recurrent STRN/ALK fusion in thyroid carcinomas. PLoS ONE 2014, 9, e87170. [Google Scholar] [CrossRef]
- Leeman-Neill, R.J.; Kelly, L.M.; Liu, P.; Brenner, A.V.; Little, M.P.; Bogdanova, T.I.; Evdokimova, V.N.; Hatch, M.; Zurnadzy, L.Y.; Nikiforova, M.N.; et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer 2014, 120, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.L.; Vyas, M.; Horne, M.J.; Virk, R.K.; Morotti, R.; Liu, Z.; Tallini, G.; Nikiforova, M.N.; Christison-Lagay, E.R.; Udelsman, R.; et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in northeast United States. Cancer 2016, 122, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Miranda, C.; Pierotti, M.A. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol. Cell. Endocrinol. 2010, 321, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Ciampi, R.; Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Melillo, R.M.; Fusco, A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur. J. Endocrinol. 2006, 155, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.D.; Zeiger, M.A. The RET oncogene in papillary thyroid carcinoma. Cancer 2015, 121, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Accardo, G.; Conzo, G.; Esposito, D.; Gambardella, C.; Mazzella, M.; Castaldo, F.; Di Donna, C.; Polistena, A.; Avenia, N.; Colantuoni, V.; et al. Genetics of medullary thyroid cancer: An overview. Int. J. Surg. 2017, 41 (Suppl. S1), S2–S6. [Google Scholar] [CrossRef]
- Mohammadi, M.; Hedayati, M. A Brief Review on the Molecular Basis of Medullary Thyroid Carcinoma. Cell J. 2017, 18, 485–492. [Google Scholar] [PubMed]
- Mulligan, L.M.; Eng, C.; Healey, C.S.; Clayton, D.; Kwok, J.B.; Gardner, E.; Ponder, M.A.; Frilling, A.; Jackson, C.E.; Lehnert, H.; et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat. Genet. 1994, 6, 70–74. [Google Scholar] [CrossRef]
- Marx, S.J. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat. Rev. Cancer 2005, 5, 367–375. [Google Scholar] [CrossRef]
- Elisei, R.; Cosci, B.; Romei, C.; Bottici, V.; Renzini, G.; Molinaro, E.; Agate, L.; Vivaldi, A.; Faviana, P.; Basolo, F.; et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: A 10-year follow-up study. J. Clin. Endocrinol. Metab. 2008, 93, 682–687. [Google Scholar] [CrossRef] [PubMed]
- St Bernard, R.; Zheng, L.; Liu, W.; Winer, D.; Asa, S.L.; Ezzat, S. Fibroblast growth factor receptors as molecular targets in thyroid carcinoma. Endocrinology 2005, 146, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Dong, J.; Xie, J.; Xing, M. Uncommon GNAQ, MMP8, AKT3, EGFR, and PIK3R1 mutations in thyroid cancers. Endocr. Pathol. 2011, 22, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Gerber, T.S.; Schad, A.; Hartmann, N.; Springer, E.; Zechner, U.; Musholt, T.J. Targeted next-generation sequencing of cancer genes in poorly differentiated thyroid cancer. Endocr. Connect. 2018, 7, 47–55. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef]
- Wu, G.; Mambo, E.; Guo, Z.; Hu, S.; Huang, X.; Gollin, S.M.; Trink, B.; Ladenson, P.W.; Sidransky, D.; Xing, M. Uncommon mutation, but common amplifications, of the PIK3CA gene in thyroid tumors. J. Clin. Endocrinol. Metab. 2005, 90, 4688–4693. [Google Scholar] [CrossRef]
- Hou, P.; Liu, D.; Shan, Y.; Hu, S.; Studeman, K.; Condouris, S.; Wang, Y.; Trink, A.; El-Naggar, A.K.; Tallini, G.; et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin. Cancer Res. 2007, 13, 1161–1170. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, P.; Yu, H.; Wang, W.; Ji, M.; Zhao, S.; Yan, S.; Sun, X.; Liu, D.; Shi, B.; et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J. Clin. Endocrinol. Metab. 2007, 92, 2387–2390. [Google Scholar] [CrossRef]
- Hou, P.; Ji, M.; Xing, M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer 2008, 113, 2440–2447. [Google Scholar] [CrossRef]
- Vasko, V.; Ferrand, M.; Di Cristofaro, J.; Carayon, P.; Henry, J.F.; de Micco, C. Specific pattern of RAS oncogene mutations in follicular thyroid tumors. J. Clin. Endocrinol. Metab. 2003, 88, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J.; et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 2013, 98, E364–E369. [Google Scholar] [CrossRef] [PubMed]
- DeLellis, R.A. Pathology and genetics of thyroid carcinoma. J. Surg. Oncol. 2006, 94, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- De Biase, D.; Cesari, V.; Visani, M.; Casadei, G.P.; Cremonini, N.; Gandolfi, G.; Sancisi, V.; Ragazzi, M.; Pession, A.; Ciarrocchi, A.; et al. High-sensitivity BRAF mutation analysis: BRAF V600E is acquired early during tumor development but is heterogeneously distributed in a subset of papillary thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E1530–E1538. [Google Scholar] [CrossRef] [PubMed]
- Knauf, J.A.; Sartor, M.A.; Medvedovic, M.; Lundsmith, E.; Ryder, M.; Salzano, M.; Nikiforov, Y.E.; Giordano, T.J.; Ghossein, R.A.; Fagin, J.A. Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFbeta signaling. Oncogene 2011, 30, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Torregrossa, L.; Viola, D.; Sensi, E.; Giordano, M.; Piaggi, P.; Romei, C.; Materazzi, G.; Miccoli, P.; Elisei, R.; Basolo, F. Papillary Thyroid Carcinoma With Rare Exon 15 BRAF Mutation Has Indolent Behavior: A Single-Institution Experience. J. Clin. Endocrinol. Metab. 2016, 101, 4413–4420. [Google Scholar] [CrossRef]
- Ciampi, R.; Knauf, J.A.; Kerler, R.; Gandhi, M.; Zhu, Z.; Nikiforova, M.N.; Rabes, H.M.; Fagin, J.A.; Nikiforov, Y.E. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J. Clin. Investig. 2005, 115, 94–101. [Google Scholar] [CrossRef]
- Sastre-Perona, A.; Santisteban, P. Role of the wnt pathway in thyroid cancer. Front. Endocrinol (Lausanne) 2012, 3, 31. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Camp, R.L.; Herrero, A.; Carcangiu, M.L.; Rimm, D.L.; Tallini, G. Beta-catenin dysregulation in thyroid neoplasms: Down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am. J. Pathol. 2001, 158, 987–996. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Tallini, G.; Herrero, A.; D’Aquila, T.G.; Carcangiu, M.L.; Rimm, D.L. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999, 59, 1811–1815. [Google Scholar] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Manzella, L.; Stella, S.; Pennisi, M.S.; Tirro, E.; Massimino, M.; Romano, C.; Puma, A.; Tavarelli, M.; Vigneri, P. New Insights in Thyroid Cancer and p53 Family Proteins. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Karunamurthy, A.; Panebianco, F.; Hsiao, S.J.; Vorhauer, J.; Nikiforova, M.N.; Chiosea, S.; Nikiforov, Y.E. Prevalence and phenotypic correlations of EIF1AX mutations in thyroid nodules. Endocr. Relat. Cancer 2016, 23, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Hemerly, J.P.; Bastos, A.U.; Cerutti, J.M. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur. J. Endocrinol. 2010, 163, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Bojdani, E.; Xing, M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem. Biophys. Res. Commun. 2010, 393, 555–559. [Google Scholar] [CrossRef]
- Sauer, S. Ligands for the Nuclear Peroxisome Proliferator-Activated Receptor Gamma. Trends Pharmacol. Sci. 2015, 36, 688–704. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, S.; Margeli, A.; Vielh, P.; Kouraklis, G. Peroxisome proliferator-activated receptor-gamma ligands as cell-cycle modulators. Cancer Treat. Rev. 2004, 30, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, N.L.; Grebe, S.K.; McIver, B.; Reddi, H.V. The role of the PAX8/PPARgamma fusion oncogene in the pathogenesis of follicular thyroid cancer. Mol. Cell. Endocrinol. 2010, 321, 50–56. [Google Scholar] [CrossRef]
- Raman, P.; Koenig, R.J. Pax-8-PPAR-gamma fusion protein in thyroid carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.; da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qu, S.; Liu, R.; Sheng, C.; Shi, X.; Zhu, G.; Murugan, A.K.; Guan, H.; Yu, H.; Wang, Y.; et al. TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, E1130–E1136. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Alexandrou, P.; Delladetsima, I.; Giannopoulou, I.; Patsouris, E.; Theocharis, S. Clinical significance of histone deacetylase (HDAC)-1, HDAC-2, HDAC-4, and HDAC-6 expression in human malignant and benign thyroid lesions. Tumour Biol. 2014, 35, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Niu, W.; Du, F.; Du, C.; Li, S.; Wang, J.; Li, L.; Wang, F.; Hao, Y.; Li, C.; et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J. Hematol. Oncol. 2016, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Du, F.; Gao, M.; Ji, Q.; Li, Z.; Zhang, Y.; Guo, Z.; Wang, J.; Chen, X.; Wang, J.; et al. Anlotinib for the Treatment of Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer. Thyroid 2018, 28, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tang, P.Z.; Chen, X.; Ge, M.; Zhang, Y.; Guo, Z.; Wang, J.; Shi, F.; Zhang, J.; Cheng, Y.; et al. Anlotinib treatment in locally advanced or metastatic medullary thyroid carcinoma: A multicenter, randomized, double-blind, placebo-controlled phase IIB trial. J. Clin. Oncol. 2019, 37, 6019. [Google Scholar] [CrossRef]
- Rugo, H.S.; Herbst, R.S.; Liu, G.; Park, J.W.; Kies, M.S.; Steinfeldt, H.M.; Pithavala, Y.K.; Reich, S.D.; Freddo, J.L.; Wilding, G. Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: Pharmacokinetic and clinical results. J. Clin. Oncol. 2005, 23, 5474–5483. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.; Rosen, L.S.; Vokes, E.E.; Kies, M.S.; Forastiere, A.A.; Worden, F.P.; Kane, M.A.; Sherman, E.; Kim, S.; Bycott, P.; et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: Results from a phase II study. J. Clin. Oncol. 2008, 26, 4708–4713. [Google Scholar] [CrossRef] [PubMed]
- Locati, L.D.; Licitra, L.; Agate, L.; Ou, S.H.; Boucher, A.; Jarzab, B.; Qin, S.; Kane, M.A.; Wirth, L.J.; Chen, C.; et al. Treatment of advanced thyroid cancer with axitinib: Phase 2 study with pharmacokinetic/pharmacodynamic and quality-of-life assessments. Cancer 2014, 120, 2694–2703. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef]
- Kurzrock, R.; Sherman, S.I.; Ball, D.W.; Forastiere, A.A.; Cohen, R.B.; Mehra, R.; Pfister, D.G.; Cohen, E.E.; Janisch, L.; Nauling, F.; et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 2011, 29, 2660–2666. [Google Scholar] [CrossRef]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef]
- Schlumberger, M.; Elisei, R.; Muller, S.; Schoffski, P.; Brose, M.; Shah, M.; Licitra, L.; Krajewska, J.; Kreissl, M.C.; Niederle, B.; et al. Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann. Oncol. 2017, 28, 2813–2819. [Google Scholar] [CrossRef]
- Traynor, K. Cabozantinib approved for advanced medullary thyroid cancer. Am. J. Health Syst. Pharm. 2013, 70, 88. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; de Souza, J.A.; Geyer, S.; Wirth, L.J.; Menefee, M.E.; Liu, S.V.; Shah, K.; Wright, J.; Shah, M.H. Cabozantinib As Salvage Therapy for Patients With Tyrosine Kinase Inhibitor-Refractory Differentiated Thyroid Cancer: Results of a Multicenter Phase II International Thyroid Oncology Group Trial. J. Clin. Oncol. 2017, 35, 3315–3321. [Google Scholar] [CrossRef]
- Ha, H.T.; Lee, J.S.; Urba, S.; Koenig, R.J.; Sisson, J.; Giordano, T.; Worden, F.P. A phase II study of imatinib in patients with advanced anaplastic thyroid cancer. Thyroid 2010, 20, 975–980. [Google Scholar] [CrossRef]
- Frank-Raue, K.; Fabel, M.; Delorme, S.; Haberkorn, U.; Raue, F. Efficacy of imatinib mesylate in advanced medullary thyroid carcinoma. Eur. J. Endocrinol. 2007, 157, 215–220. [Google Scholar] [CrossRef]
- De Groot, J.W.; Zonnenberg, B.A.; van Ufford-Mannesse, P.Q.; de Vries, M.M.; Links, T.P.; Lips, C.J.; Voest, E.E. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2007, 92, 3466–3469. [Google Scholar] [CrossRef]
- Okamoto, K.; Kodama, K.; Takase, K.; Sugi, N.H.; Yamamoto, Y.; Iwata, M.; Tsuruoka, A. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion-driven tumor models. Cancer Lett. 2013, 340, 97–103. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Schlumberger, M.; Jarzab, B.; Martins, R.G.; Pacini, F.; Robinson, B.; McCaffrey, J.C.; Shah, M.H.; Bodenner, D.L.; Topliss, D.; et al. A phase 2 trial of lenvatinib (E7080) in advanced, progressive, radioiodine-refractory, differentiated thyroid cancer: A clinical outcomes and biomarker assessment. Cancer 2015, 121, 2749–2756. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef]
- Haddad, R.I.; Nasr, C.; Bischoff, L.; Busaidy, N.L.; Byrd, D.; Callender, G.; Dickson, P.; Duh, Q.Y.; Ehya, H.; Goldner, W.; et al. NCCN Guidelines Insights: Thyroid Carcinoma, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 1429–1440. [Google Scholar] [CrossRef]
- Schlumberger, M.; Jarzab, B.; Cabanillas, M.E.; Robinson, B.; Pacini, F.; Ball, D.W.; McCaffrey, J.; Newbold, K.; Allison, R.; Martins, R.G.; et al. A Phase II Trial of the Multitargeted Tyrosine Kinase Inhibitor Lenvatinib (E7080) in Advanced Medullary Thyroid Cancer. Clin. Cancer Res. 2016, 22, 44–53. [Google Scholar] [CrossRef]
- Polverino, A.; Coxon, A.; Starnes, C.; Diaz, Z.; DeMelfi, T.; Wang, L.; Bready, J.; Estrada, J.; Cattley, R.; Kaufman, S.; et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res. 2006, 66, 8715–8721. [Google Scholar] [CrossRef]
- Sherman, S.I.; Wirth, L.J.; Droz, J.P.; Hofmann, M.; Bastholt, L.; Martins, R.G.; Licitra, L.; Eschenberg, M.J.; Sun, Y.N.; Juan, T.; et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N. Engl. J. Med. 2008, 359, 31–42. [Google Scholar] [CrossRef]
- Schlumberger, M.J.; Elisei, R.; Bastholt, L.; Wirth, L.J.; Martins, R.G.; Locati, L.D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.P.; et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J. Clin. Oncol. 2009, 27, 3794–3801. [Google Scholar] [CrossRef]
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol. Cancer Ther. 2007, 6, 2012–2021. [Google Scholar] [CrossRef]
- Bible, K.C.; Suman, V.J.; Molina, J.R.; Smallridge, R.C.; Maples, W.J.; Menefee, M.E.; Rubin, J.; Sideras, K.; Morris, J.C., 3rd; McIver, B.; et al. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: Results of a phase 2 consortium study. Lancet Oncol. 2010, 11, 962–972. [Google Scholar] [CrossRef]
- Bible, K.C.; Suman, V.J.; Menefee, M.E.; Smallridge, R.C.; Molina, J.R.; Maples, W.J.; Karlin, N.J.; Traynor, A.M.; Kumar, P.; Goh, B.C.; et al. A multiinstitutional phase 2 trial of pazopanib monotherapy in advanced anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 2012, 97, 3179–3184. [Google Scholar] [CrossRef]
- Bible, K.C.; Suman, V.J.; Molina, J.R.; Smallridge, R.C.; Maples, W.J.; Menefee, M.E.; Rubin, J.; Karlin, N.; Sideras, K.; Morris, J.C., 3rd; et al. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J. Clin. Endocrinol. Metab. 2014, 99, 1687–1693. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Schneider, T.C.; Abdulrahman, R.M.; Corssmit, E.P.; Morreau, H.; Smit, J.W.; Kapiteijn, E. Long-term analysis of the efficacy and tolerability of sorafenib in advanced radio-iodine refractory differentiated thyroid carcinoma: Final results of a phase II trial. Eur. J. Endocrinol. 2012, 167, 643–650. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Lam, E.T.; Ringel, M.D.; Kloos, R.T.; Prior, T.W.; Knopp, M.V.; Liang, J.; Sammet, S.; Hall, N.C.; Wakely, P.E., Jr.; Vasko, V.V.; et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J. Clin. Oncol. 2010, 28, 2323–2330. [Google Scholar] [CrossRef]
- Savvides, P.; Nagaiah, G.; Lavertu, P.; Fu, P.; Wright, J.J.; Chapman, R.; Wasman, J.; Dowlati, A.; Remick, S.C. Phase II trial of sorafenib in patients with advanced anaplastic carcinoma of the thyroid. Thyroid 2013, 23, 600–604. [Google Scholar] [CrossRef]
- Chow, L.Q.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef]
- Carr, L.L.; Mankoff, D.A.; Goulart, B.H.; Eaton, K.D.; Capell, P.T.; Kell, E.M.; Bauman, J.E.; Martins, R.G. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin. Cancer Res. 2010, 16, 5260–5268. [Google Scholar] [CrossRef]
- Bikas, A.; Kundra, P.; Desale, S.; Mete, M.; O’Keefe, K.; Clark, B.G.; Wray, L.; Gandhi, R.; Barett, C.; Jelinek, J.S.; et al. Phase 2 clinical trial of sunitinib as adjunctive treatment in patients with advanced differentiated thyroid cancer. Eur. J. Endocrinol. 2016, 174, 373–380. [Google Scholar] [CrossRef]
- Ravaud, A.; de la Fouchardiere, C.; Caron, P.; Doussau, A.; Do Cao, C.; Asselineau, J.; Rodien, P.; Pouessel, D.; Nicolli-Sire, P.; Klein, M.; et al. A multicenter phase II study of sunitinib in patients with locally advanced or metastatic differentiated, anaplastic or medullary thyroid carcinomas: Mature data from the THYSU study. Eur. J. Cancer 2017, 76, 110–117. [Google Scholar] [CrossRef]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002, 62, 7284–7290. [Google Scholar] [PubMed]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar] [PubMed]
- Wells, S.A., Jr.; Gosnell, J.E.; Gagel, R.F.; Moley, J.; Pfister, D.; Sosa, J.A.; Skinner, M.; Krebs, A.; Vasselli, J.; Schlumberger, M. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Oncol. 2010, 28, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Thornton, K.; Kim, G.; Maher, V.E.; Chattopadhyay, S.; Tang, S.; Moon, Y.J.; Song, P.; Marathe, A.; Balakrishnan, S.; Zhu, H.; et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: U.S. Food and Drug Administration drug approval summary. Clin. Cancer Res. 2012, 18, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Bastholt, L.; Krause, T.; de la Fouchardiere, C.; Tennvall, J.; Awada, A.; Gomez, J.M.; Bonichon, F.; Leenhardt, L.; Soufflet, C.; et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 2 trial. Lancet Oncol. 2012, 13, 897–905. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, C.; Gao, W.; Cui, R.; Liang, J. Overwhelming rapid metabolic and structural response to apatinib in radioiodine refractory differentiated thyroid cancer. Oncotarget 2017, 8, 42252–42261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, C.; Lin, Y. Pilot Dose Comparison of Apatinib in Chinese Patients With Progressive Radioiodine-Refractory Differentiated Thyroid Cancer. J. Clin. Endocrinol. Metab. 2018, 103, 3640–3646. [Google Scholar] [CrossRef]
- Falchook, G.S.; Millward, M.; Hong, D.; Naing, A.; Piha-Paul, S.; Waguespack, S.G.; Cabanillas, M.E.; Sherman, S.I.; Ma, B.; Curtis, M.; et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid 2015, 25, 71–77. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gao, Q.; Fu, M.; Ni, N.; Pei, Y.; Ou, W.B. Anaplastic lymphoma kinase fusions: Roles in cancer and therapeutic perspectives. Oncol. Lett. 2019, 17, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wolfstetter, G.; Siaw, J.; Chand, D.; Hugosson, F.; Palmer, R.H.; Hallberg, B. Anaplastic lymphoma kinase L1198F and G1201E mutations identified in anaplastic thyroid cancer patients are not ligand-independent. Oncotarget 2017, 8, 11566–11578. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.S.; Beck, J.T.; Edenfield, W.J.; et al. Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): A phase 1b open-label study. Am. J. Hematol. 2018, 93, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Godbert, Y.; Henriques de Figueiredo, B.; Bonichon, F.; Chibon, F.; Hostein, I.; Perot, G.; Dupin, C.; Daubech, A.; Belleannee, G.; Gros, A.; et al. Remarkable Response to Crizotinib in Woman With Anaplastic Lymphoma Kinase-Rearranged Anaplastic Thyroid Carcinoma. J. Clin. Oncol. 2015, 33, e84–e87. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.H.; Oh, Y.L.; Hong, M.; Yun, J.W.; Lee, H.W.; Kim, D.; Ji, Y.; Kim, D.H.; Park, W.Y.; Shin, H.T.; et al. Identification of Driving ALK Fusion Genes and Genomic Landscape of Medullary Thyroid Cancer. PLoS Genet. 2015, 11, e1005467. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Ruiz, R.; Giovannetti, E.; Gil-Bazo, I.; Russo, A.; Passiglia, F.; Giallombardo, M.; Peeters, M.; Raez, L. Entrectinib: A potent new TRK, ROS1, and ALK inhibitor. Expert Opin. Investig. Drugs 2015, 24, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Bauer, T.M.; Lee, J.J.; Dowlati, A.; Brose, M.S.; Farago, A.F.; Taylor, M.; Shaw, A.T.; Montez, S.; Meric-Bernstam, F.; et al. Larotrectinib in adult patients with solid tumours: A multi-centre, open-label, phase I dose-escalation study. Ann. Oncol. 2019, 30, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Yeager, N.; Brewer, C.; Cai, K.Q.; Xu, X.X.; Di Cristofano, A. Mammalian target of rapamycin is the key effector of phosphatidylinositol-3-OH-initiated proliferative signals in the thyroid follicular epithelium. Cancer Res. 2008, 68, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Saji, M.; Ringel, M.D. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol. Cell. Endocrinol. 2010, 321, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Jiang, T.; Rosen, D.M.; Nelkin, B.D.; Ball, D.W. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin. Cancer Res. 2011, 17, 6482–6489. [Google Scholar] [CrossRef] [PubMed]
- Borson-Chazot, F.; Dantony, E.; Illouz, F.; Lopez, J.; Niccoli, P.; Wassermann, J.; Do Cao, C.; Leboulleux, S.; Klein, M.; Tabarin, A.; et al. Effect of Buparlisib, a Pan-Class I PI3K Inhibitor, in Refractory Follicular and Poorly Differentiated Thyroid Cancer. Thyroid 2018, 28, 1174–1179. [Google Scholar] [CrossRef]
- Lim, S.M.; Chang, H.; Yoon, M.J.; Hong, Y.K.; Kim, H.; Chung, W.Y.; Park, C.S.; Nam, K.H.; Kang, S.W.; Kim, M.K.; et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann. Oncol. 2013, 24, 3089–3094. [Google Scholar] [CrossRef]
- Schneider, T.C.; de Wit, D.; Links, T.P.; van Erp, N.P.; van der Hoeven, J.J.; Gelderblom, H.; van Wezel, T.; van Eijk, R.; Morreau, H.; Guchelaar, H.J.; et al. Beneficial Effects of the mTOR Inhibitor Everolimus in Patients with Advanced Medullary Thyroid Carcinoma: Subgroup Results of a Phase II Trial. Int. J. Endocrinol. 2015, 2015, 348124. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.C.; de Wit, D.; Links, T.P.; van Erp, N.P.; van der Hoeven, J.J.; Gelderblom, H.; Roozen, I.C.; Bos, M.; Corver, W.E.; van Wezel, T.; et al. Everolimus in Patients With Advanced Follicular-Derived Thyroid Cancer: Results of a Phase II Clinical Trial. J. Clin. Endocrinol. Metab. 2017, 102, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Hanna, G.J.; Busaidy, N.L.; Chau, N.G.; Wirth, L.J.; Barletta, J.A.; Calles, A.; Haddad, R.I.; Kraft, S.; Cabanillas, M.E.; Rabinowits, G.; et al. Genomic Correlates of Response to Everolimus in Aggressive Radioiodine-refractory Thyroid Cancer: A Phase II Study. Clin. Cancer Res. 2018, 24, 1546–1553. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Zhang, G.; Lallani, S.B.; Chen, Z.; Martinson, D.E.; Khuri, F.R.; Lonial, S.; Marcus, A.; Sun, S.Y. Evaluation of preclinical efficacy of everolimus and pasireotide in thyroid cancer cell lines and xenograft models. PLoS ONE 2019, 14, e0206309. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.; England, J.A.; Rafferty, A.; Jesudason, V.; Bedford, K.; Karsai, L.; Atkin, S.L. Somatostatin receptor expression in thyroid disease. Int. J. Exp. Pathol. 2013, 94, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Faggiano, A.; Modica, R.; Severino, R.; Camera, L.; Fonti, R.; Del Prete, M.; Chiofalo, M.G.; Aria, M.; Ferolla, P.; Vitale, G.; et al. The antiproliferative effect of pasireotide LAR alone and in combination with everolimus in patients with medullary thyroid cancer: A single-center, open-label, phase II, proof-of-concept study. Endocrine 2018, 62, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Manohar, P.M.; Beesley, L.J.; Taylor, J.M.; Hesseltine, E.; Haymart, M.R.; Esfandiari, N.H.; Hanauer, D.A.; Worden, F.P. Retrospective Study of Sirolimus and Cyclophosphamide in Patients with Advanced Differentiated Thyroid Cancers. J. Thyroid Disord. Ther. 2015, 4. [Google Scholar] [CrossRef]
- Sherman, E.J.; Dunn, L.A.; Ho, A.L.; Baxi, S.S.; Ghossein, R.A.; Fury, M.G.; Haque, S.; Sima, C.S.; Cullen, G.; Fagin, J.A.; et al. Phase 2 study evaluating the combination of sorafenib and temsirolimus in the treatment of radioactive iodine-refractory thyroid cancer. Cancer 2017, 123, 4114–4121. [Google Scholar] [CrossRef]
- Grommes, C.; Landreth, G.E.; Heneka, M.T. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004, 5, 419–429. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P.; et al. Efatutazone, an oral PPAR-gamma agonist, in combination with paclitaxel in anaplastic thyroid cancer: Results of a multicenter phase 1 trial. J. Clin. Endocrinol. Metab. 2013, 98, 2392–2400. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Haghpanah, V.; Malehmir, M.; Larijani, B.; Ahmadian, S.; Alimoghaddam, K.; Heshmat, R.; Ghavamzadeh, A.; Adabi, K.; Ghaffari, S.H. The Beneficial Effects of Valproic Acid in Thyroid Cancer Are Mediated through Promoting Redifferentiation and Reducing Stemness Level: An In Vitro Study. J. Thyroid Res. 2014, 2014, 218763. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.T.; Zheng, H.B.; Zhou, L.; Zhang, D.Q.; Liu, X.L.; Sun, H. Valproic acid, targets papillary thyroid cancer through inhibition of c-Met signalling pathway. Am. J. Transl. Res. 2017, 9, 3138–3147. [Google Scholar] [PubMed]
- Nilubol, N.; Merkel, R.; Yang, L.; Patel, D.; Reynolds, J.C.; Sadowski, S.M.; Neychev, V.; Kebebew, E. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin. Endocrinol. 2017, 86, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- French, J.D.; Weber, Z.J.; Fretwell, D.L.; Said, S.; Klopper, J.P.; Haugen, B.R. Tumor-associated lymphocytes and increased FoxP3+ regulatory T cell frequency correlate with more aggressive papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2010, 95, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Choudhury, A.; Lladser, A.; Kiessling, R.; Johansson, C.C. Regulatory T cells in cancer. Adv. Cancer Res. 2010, 107, 57–117. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Gogali, F.; Paterakis, G.; Rassidakis, G.Z.; Kaltsas, G.; Liakou, C.I.; Gousis, P.; Neonakis, E.; Manoussakis, M.N.; Liapi, C. Phenotypical analysis of lymphocytes with suppressive and regulatory properties (Tregs) and NK cells in the papillary carcinoma of thyroid. J. Clin. Endocrinol. Metab. 2012, 97, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Veyhl, J.; Jessa, F.; Polyakova, O.; Alenzi, A.; MacMillan, C.; Ralhan, R.; Walfish, P.G. Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 2016, 7, 32318–32328. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.W.; Gigliotti, B.J.; Pai, S.I.; Parangi, S.; Wachtel, H.; Mino-Kenudson, M.; Gunda, V.; Faquin, W.C. PD-L1 and IDO1 Are Expressed in Poorly Differentiated Thyroid Carcinoma. Endocr. Pathol. 2018, 29, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Guo, T.; Huang, X.; Wu, Q.; Niu, D.; Ji, X.; Feng, Q.; Li, Z.; Kakudo, K. In papillary thyroid carcinoma, expression by immunohistochemistry of BRAF V600E, PD-L1, and PD-1 is closely related. Virchows Arch. 2018, 472, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.L.; Qu, N.; Luo, T.X.; Xiang, J.; Liao, T.; Sun, G.H.; Wang, Y.; Wang, Y.L.; Huang, C.P.; Ji, Q.H. Programmed Death-Ligand 1 Expression in Papillary Thyroid Cancer and Its Correlation with Clinicopathologic Factors and Recurrence. Thyroid 2017, 27, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Wei, W.J.; Song, H.J.; Sun, Z.K.; Shen, C.T.; Zhang, X.Y.; Chen, X.Y.; Qiu, Z.L.; Luo, Q.Y. Programmed Cell Death-Ligand 1 Overexpression in Thyroid Cancer. Endocr. Pract. 2019, 25, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.C.; Prawira, A.; de Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef]
- Sherman, E.J.; Tsai, C.J.; Zhi, W.I.; Fetten, J.V.; Wu, V.; Ho, A.L.; Riaz, N.; Pfister, D.G.; Lee, N.Y. Pilot study combining PD-L1 antibody durvalumab (D) with CTLA-4 antibody tremelimumab (T) and stereotactic body radiotherapy (SBRT) to treat metastatic anaplastic thyroid cancer (ATC). J. Clin. Oncol. 2019, 37, 6088. [Google Scholar] [CrossRef]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Massimino, M.; Stella, S.; Tirro, E.; Romano, C.; Pennisi, M.S.; Puma, A.; Manzella, L.; Zanghi, A.; Stagno, F.; Di Raimondo, F.; et al. Non ABL-directed inhibitors as alternative treatment strategies for chronic myeloid leukemia. Mol. Cancer 2018, 17, 56. [Google Scholar] [CrossRef]
- Pirosa, M.C.; Leotta, S.; Cupri, A.; Stella, S.; Martino, E.A.; Scalise, L.; Sapienza, G.; Calafiore, V.; Mauro, E.; Spadaro, A.; et al. Long-Term Molecular Remission Achieved by Antibody Anti-CD22 and Ponatinib in a Patient Affected by Ph’+ Acute Lymphoblastic Leukemia Relapsed after Second Allogeneic Hematopoietic Stem Cell Transplantation: A Case Report. Chemotherapy 2018, 63, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Tirro, E.; Massimino, M.; Romano, C.; Pennisi, M.S.; Stella, S.; Vitale, S.R.; Fidilio, A.; Manzella, L.; Parrinello, N.L.; Stagno, F.; et al. Chk1 Inhibition Restores Inotuzumab Ozogamicin Citotoxicity in CD22-Positive Cells Expressing Mutant p53. Front. Oncol. 2019, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Stagno, F.; Vigneri, P.; Del Fabro, V.; Stella, S.; Restuccia, N.; Giallongo, C.; Massimino, M.; Berretta, S.; Pennisi, M.S.; Tibullo, D.; et al. Concomitant and feasible treatment with dasatinib and the anti-EGFR antibody cetuximab plus radiotherapy in a CML patient with multiple squamous neoplasias. Acta Oncol. 2010, 49, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Bible, K.C.; Chow, L.Q.M.; Gilbert, J.; Grande, C.; Worden, F.; Haddad, R. Management of treatment-related toxicities in advanced medullary thyroid cancer. Cancer Treat. Rev. 2018, 66, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Carneiro, B.A.; Chandra, S.; Pai, S.G.; Chae, Y.K.; Kaplan, J.B.; Garrett, H.B.; Agulnik, M.; Kopp, P.A.; Giles, F.J. Spotlight on lenvatinib in the treatment of thyroid cancer: Patient selection and perspectives. Drug Des. Dev. Ther. 2016, 10, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AGENT | COMBINATION | STUDY POPULATION | DESIGN | PATIENTS | PRIMARY END POINT | STATUS | IDENTIFIER |
|---|---|---|---|---|---|---|---|
| Cabozantinib Lenvatinib Sorafenib Vandetanib | Advanced TC | Nonrandomized, Open label, phase II | 45 | PFS, OS, DLTs | Recruiting | NCT03630120 | |
| Cabozantinib | RAIR DTC pretreated with anti-VEGFR | Randomized, Double blind, phase III | 300 | PFS, ORR | Recruiting | NCT03690388 | |
| Cabozantinib | RAIR DTC | Nonrandomized, Open label, phase II | 43 | SP | Active, not recruiting | NCT02041260 | |
| Cabozantinib | Ipilimumab Nivolumab | RAIR DTC pretreated with anti-VEGFR | Nonrandomized, Open label, phase II | 24 | ORR | Not yet recruiting | NCT03914300 |
| Cabozantinib | Atezolizumab | LA or M+ Solid Tumors | Nonrandomized, Open label, phase Ib | 1000 | DLTs, ORR | Recruiting | NCT03170960 |
| Cabozantinib | Recurrent or Refractory Solid Tumors | Nonrandomized, Open label, phase I | 41 | DLTs | Active, not recruiting | NCT01709435 | |
| Imatinib | RAIR PTC | Nonrandomized, Open label, phase I | 18 | EP | Recruiting | NCT03469011 | |
| Lenvatinib | Advanced RAIR TC | Randomized, Double blind, phase II | 152 | ORR, SP | Recruiting | NCT02702388 | |
| Lenvatinib | ATC | Nonrandomized, Open label, phase II | 34 | ORR | Terminated | NCT02657369 | |
| Lenvatinib | ATC | Nonrandomized, Open label, phase II | 39 * | OS | Active, not recruiting | NCT02726503 | |
| Lenvatinib | Denosumab | Bone M+ RAIR DTC | Nonrandomized, Open label, phase II | 35 | EP | Not yet recruiting | NCT03732495 |
| Lenvatinib | RAI | RAI-sensitive DTC | Nonrandomized, Open label, phase II | 30 | PFS | Recruiting | NCT03506048 |
| Lenvatinib | Pembrolizumab | RAIR DTC | Nonrandomized, Open label, phase II | 60 | ORR | Recruiting | NCT02973997 |
| Pazopanib | TC | Randomized, Open label, phase II | 168 | PFS | Recruiting | NCT01813136 | |
| Pazopanib | Advanced TC | Nonrandomized, Open label, phase II | 152 | ORR | Active, not recruiting | NCT00625846 | |
| Pazopanib | Paclitaxel RT | ATC | Randomized, Open label, phase II | 121 * | OS, DLTs | Active, not recruiting | NCT01236547 |
| Sorafenib | Adjuvant after RAI | Nonrandomized, Open label, phase II | 32 | ORR | Completed | NCT00887107 | |
| Sorafenib | Advanced.TC | Nonrandomized, Open label, phase II | 61 | ORR | Completed | NCT00654238 | |
| Sorafenib | Advanced.TC | Nonrandomized, Open label, phase II | 25 | ORR | Terminated | NCT00095693 | |
| Vandetanib | Hereditary MTC | Nonrandomized, Open label, phase I/II | 17 | SP | Active, not recruiting | NCT00514046 | |
| Vandetanib | Advanced MTC | Randomized, Double blind, phase III | 437 | PFS | Active, not recruiting | NCT00410761 |
| TARGET | AGENT | COMBINATION | STUDY POPULATION | DESIGN | PATIENTS | PRIMARY END POINT | STATUS | IDENTIFIER |
|---|---|---|---|---|---|---|---|---|
| ALK | Alectinib | RET-rearranged NSCLC or RET-mut TC | Non-Randomized, Open Label, Phase I/II | 78* | MTD, ORR | Active, not recruiting | NCT03131206 | |
| Ceritinib | M+ or LA ATC | Single Group Assignment, Open Label, Phase II | 100* | Development of progression | Recruiting | NCT02289144 | ||
| BRAF | Dabrafenib | Trametinib | Recurrent TC | Randomized, Open label, phase II | 53 | ORR | Active, not recruiting | NCT01723202 |
| Dabrafenib | Trametinib RAI | M+ RAIR with RAS or BRAF mutation | Nonrandomized, Open label, phase II | 87 | ORR | Recruiting | NCT03244956 | |
| Dabrafenib | Lapatinib | TC with BRAF mutation | Nonrandomized, Open label, phase I | 18 | DLTs | Active, not recruiting | NCT01947023 | |
| Vemurafenib | Neoadjuvant-Advanced TC | Nonrandomized, Open label, phase II | 24 | EP | Active, not recruiting | NCT01709292 | ||
| MEK | Selumetinib | Olaparib | Solid tumors with Ras pathway alterations, and ovarian tumors with PARP resistance | Nonrandomized, Open label, phase I | 90 | DLTs | Recruiting | NCT03162627 |
| Selumetinib | 131I | Recurrent or M+ TC | Randomized, Double blind, phase II | 60 | ORR | Recruiting | NCT02393690 | |
| Trametinib | Paclitaxel | Advanced ATC | Nonrandomized, Open label, phase I | 12 | PFS | Recruiting | NCT03085056 | |
| Trametinib | Pazopanib | Advanced Solid Tumors (DTC, STS and Chol) | Nonrandomized, Open label, phase I | 89 | DLTs, SP | Completed | NCT01438554 | |
| Trametinib | RAI | RAS mutant or RAS/RAF wild-type, RAIR and/or M+ TC | Nonrandomized, Open label, phase II | 35 | PFS, ORR | Recruiting | NCT02152995 | |
| NTRK | Entrectinib | LA or M+ Solid Tumors harboring NTRK1/2/3, ROS1, or ALK Rearrangements | Non-Randomized, Open Label, Phase II | 300* | ORR | Recruiting | NCT02568267 | |
| Entrectinib | Solid tumors with or without TRK, ROS1 or ALK Fusions | Non-Randomized, Open label, Phase I | 65* | MTD, RP2D, ORR | Recruiting | NCT02650401 | ||
| Larotrectinib | Solid Tumors Harboring NTRK Fusion | Non-Randomized, Open Label, Phase II | 320* | ORR | Recruiting | NCT02576431 | ||
| LOXO-195 | Patients with previously treated NTRK Fusion cancers | Single Group Assignment, Open label, Phase I/II | 93* | MTD, recommended dose, PR, CR | Recruiting | NCT03215511 | ||
| PPAR-γ | Efatutazone | Paclitaxel | Advanced ATC | Nonrandomized, Open label, phase II | 19 | ORR | Active, not Recruiting | NCT02152137 |
| VEGFR-2 | Apatinib | RT | Inoperable or RAIR TC | Nonrandomized, Open label, phase II | 20 | PFS | Recruiting | NCT03300765 |
| Apatinib | RAIR DTC | Randomized, Double blind, phase III | 118 | PFS | Recruiting | NCT03048877 | ||
| Apatinib | Locally Advanced/M+ DTC | Nonrandomized, Open label, phase II | 20 | EP | Recruiting | NCT03167385 | ||
| Apatinib | Local Progressive/M+ RAIR | Nonrandomized, Open label, phase II | 40 | ORR | Recruiting | NCT03199677 |
| AGENT | COMBINATION | STUDY POPULATION | DESIGN | PATIENTS | PRIMARY END POINT | STATUS | IDENTIFIER |
|---|---|---|---|---|---|---|---|
| Everolimus | LA or M+ TC | Nonrandomized, Open label, phase II | 40 | ORR | Completed | NCT01164176 | |
| Everolimus | RAIR TC | Nonrandomized, Open label, phase II | 33 | PFS | Active, not Recruiting | NCT00936858 | |
| Everolimus | Lenvatinib | M+ DTC progressed on Lenvatinib alone | Nonrandomized, Open label, phase II | 40 | PFS | Recruiting | NCT03139747 |
| Everolimus | Neratinib | Advanced Cancer with EGFR/HER2 Mut/Ampl, HER3/4 Mut | Nonrandomized, Open label, phase I | 120 | DLTs | Recruiting | NCT03065387 |
| Everolimus | Pasireotide | RAIR DTC and MTC | Randomized, Open label, phase II | 42 | ORR | Completed | NCT01270321 |
| Everolimus | Sorefenib | M+ DTC progressed on Sorafenib alone | Nonrandomized, Open label, phase II | 40 | ORR, PFS | Active, not Recruiting | NCT01263951 |
| Everolimus | Sorefenib | Advanced TC never treated with m-TOR inhibitor or Sorafenib | Nonrandomized, Open label, phase II | 41 | ORR | Active, not Recruiting | NCT01141309 |
| Everolimus | Sorefenib | Advanced RAIR Hurthle Cell TC | Randomized, Open label, phase II | 34l | PFS | Recruiting | NCT02143726 |
| Everolimus | Vatalinib | Advanced Solid Tumors | Nonrandomized, Open label, phase I | 96 | DLTs, SP | Completed | NCT00655655 |
| Sirolimus | Ciclofosfamide | M+ or RAIR DTC | Nonrandomized, Open label, phase II | 19 | ORR | Recruiting | NCT03099356 |
| Sirolimus | Grapefruit juice | Advanced Malignancies | Nonrandomized, Open label, phase Ib | 41 | PK | Completed | NCT00375245 |
| Temsirolimus | Bevacizumab Valproic Acid | Advanced or M+ Malignancy or Other Benign Disease | Nonrandomized, Open label, phase I | 216 | DLTs | Recruiting | NCT01552434 |
| Temsirolimus | Vinorelbine | Unresectable or M+ Solid Tumors | Nonrandomized, Open label, phase I | 19 | DLTs, ORR | Completed | NCT01155258 |
| TARGET | AGENT | COMBINATION | STUDY POPULATION | DESIGN | PATIENTS | PRIMARY ENDPOINT | STATUS | IDENTIFIER |
|---|---|---|---|---|---|---|---|---|
| PD-1 | Pembrolizumab | M+ or LA ATC | Single Group Assignment Open label, phase II | 20 | RR | Recruiting | NCT02688608 | |
| Pembrolizumab | Recurrent or M+ MTC | Nonrandomized Parallel Assignment Open label, phase II | 32 | DLTs | Recruiting | NCT03072160 | ||
| Pembrolizumab | Patients with rare cancer types | Single Group Assignment, Open label, phase II | 350 | ORR | Recruiting | NCT03012620 | ||
| Pembrolizumab | Advanced Solid Tumors | Single Group Assignment, Open label, phase II | 1350 | ORR | Recruiting | NCT02628067 | ||
| Pembrolizumab | Docetaxel | Poorly Chemo-responsive Thyroid and Salivary Gland Tumors | Nonrandomized, Parallel Assignment Open label, phase I | 46 | RR | Recruiting | NCT03360890 | |
| Pembrolizumab | Docetaxel Doxorubicin | ATC | Nonrandomized, Open label, phase II | 3* | OSR | Active, not Recruiting | NCT03211117 | |
| Pembrolizumab | Lenvatinib | RAIR DTC | Single Group Assignment, Open label, phase II | 60 | CRR | Recruiting | NCT02973997 | |
| PD-1 and CTLA-4 | Nivolumab Ipilimumab | RAIR DTC, ATC, MTC | Randomized, Parallel Assignment Open label, phase II | 54 | RRR | Recruiting | NCT03246958 | |
| PD-L1 | Atezolizumab | Bevacizumab Cobimetinib Paclitaxel Vemurafenib | ATC, PDTC | Nonrandomized, Parallel Assignment Open label, phase II | 50 | OS | Recruiting | NCT03181100 |
| Atezolizumab | Cabozantinib | M+ ATC | Nonrandomized Sequential Assignment Open label, phase I-II | 1000 | DLTs | Recruiting | NCT03170960 | |
| Durvalumab | TC | Single Group Assignment Open label, phase I | 11 | DLTs | Recruiting | NCT03215095 | ||
| PD-L1 and CTLA-4 | Durvalumab+ Tremelimumab | M+ ATC | Single Group Assignment Open label, phase | 13 | OS | Active, not Recruiting | NCT03122496 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirrò, E.; Martorana, F.; Romano, C.; Vitale, S.R.; Motta, G.; Di Gregorio, S.; Massimino, M.; Pennisi, M.S.; Stella, S.; Puma, A.; et al. Molecular Alterations in Thyroid Cancer: From Bench to Clinical Practice. Genes 2019, 10, 709. https://doi.org/10.3390/genes10090709
Tirrò E, Martorana F, Romano C, Vitale SR, Motta G, Di Gregorio S, Massimino M, Pennisi MS, Stella S, Puma A, et al. Molecular Alterations in Thyroid Cancer: From Bench to Clinical Practice. Genes. 2019; 10(9):709. https://doi.org/10.3390/genes10090709
Chicago/Turabian StyleTirrò, Elena, Federica Martorana, Chiara Romano, Silvia Rita Vitale, Gianmarco Motta, Sandra Di Gregorio, Michele Massimino, Maria Stella Pennisi, Stefania Stella, Adriana Puma, and et al. 2019. "Molecular Alterations in Thyroid Cancer: From Bench to Clinical Practice" Genes 10, no. 9: 709. https://doi.org/10.3390/genes10090709
APA StyleTirrò, E., Martorana, F., Romano, C., Vitale, S. R., Motta, G., Di Gregorio, S., Massimino, M., Pennisi, M. S., Stella, S., Puma, A., Gianì, F., Russo, M., Manzella, L., & Vigneri, P. (2019). Molecular Alterations in Thyroid Cancer: From Bench to Clinical Practice. Genes, 10(9), 709. https://doi.org/10.3390/genes10090709