Network as a Biomarker: A Novel Network-Based Sparse Bayesian Machine for Pathway-Driven Drug Response Prediction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
Sparse Bayesian Classifier Combined with Disease-Specific Network
3. Results
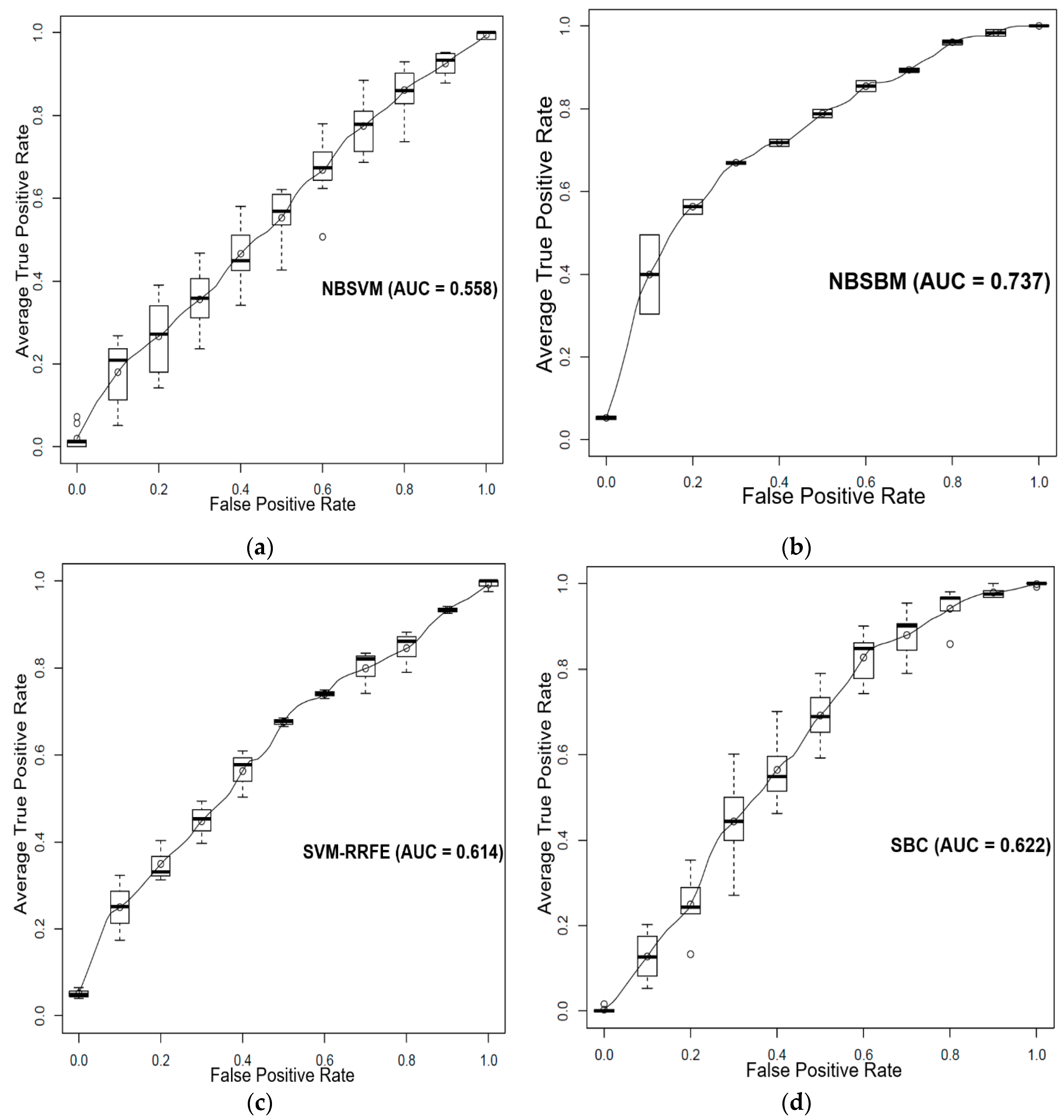
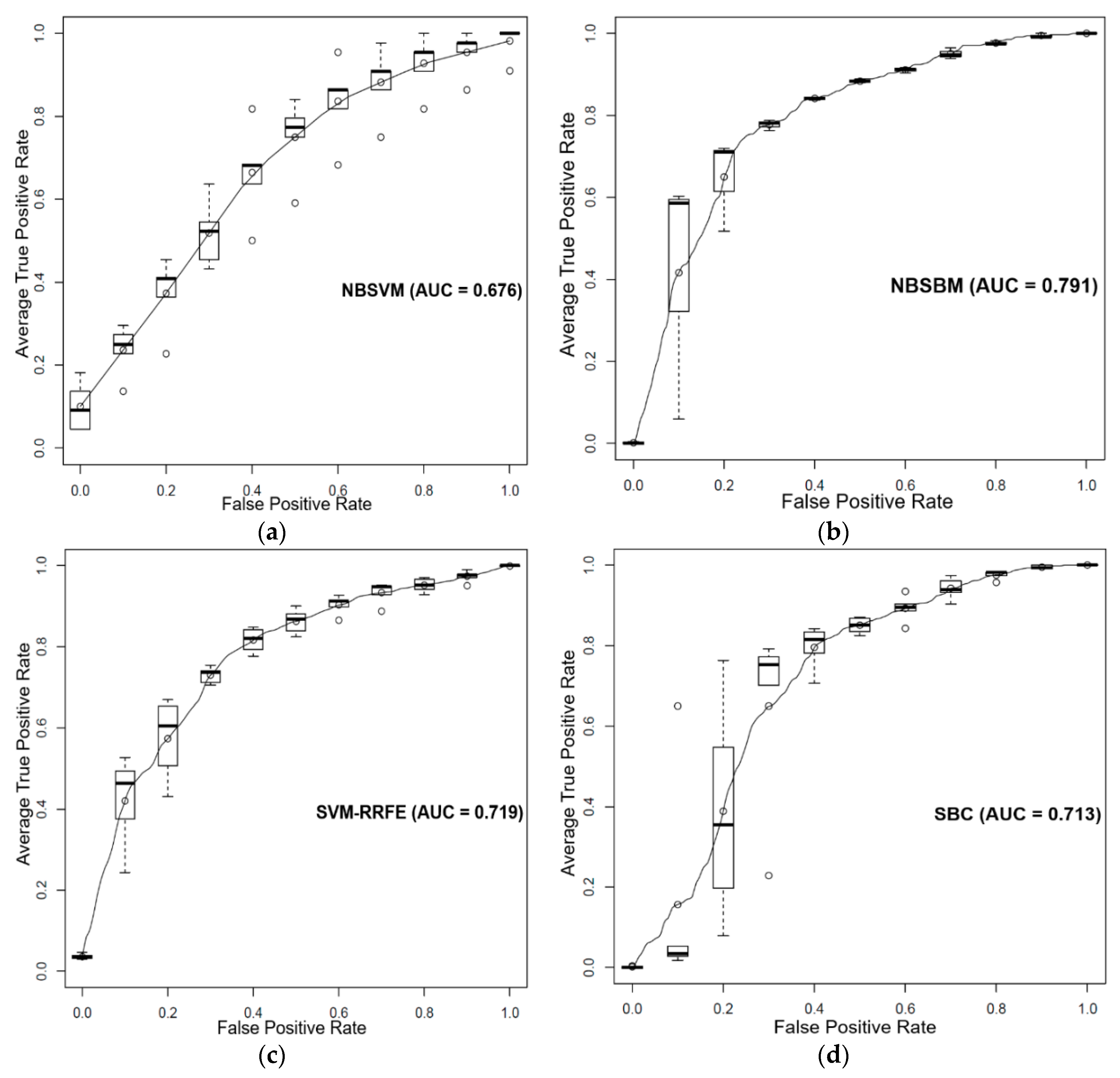
3.1. Prediction of Sensitivity and Resistance of Prostate Cancer Cell Lines to Dasatinib
3.2. Prediction of Sensitivity and Resistance of Breast Cancer Patients to Tamoxifen
3.3. Prediction of Sensitivity and Resistance of Various Cancer Cells to Dasatinib
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sen, B.; Peng, S.; Tang, X.; Erickson, H.S.; Galindo, H.; Mazumdar, T.; Stewart, D.J.; Wistuba, I.; Johnson, F.M. Kinase-Impaired BRAF Mutations in Lung Cancer Confer Sensitivity to Dasatinib. Sci. Transl. Med. 2012, 4, 136ra70. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-D.; Reeves, K.; Luo, F.R.; Xu, L.-A.; Lee, F.; Clark, E.; Huang, F. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: Rationale for patient selection and efficacy monitoring. Genome Biol. 2007, 8, R255. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Reeves, K.; Han, X.; Fairchild, C.; Platero, S.; Wong, T.W.; Lee, F.; Shaw, P.; Clark, E. Identification of Candidate Molecular Markers Predicting Sensitivity in Solid Tumors to Dasatinib: Rationale for Patient Selection. Cancer Res. 2007, 67, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, H. Network-constrained regularization and variable selection for analysis of genomic data. Bioinformatics 2008, 24, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, F.; Zinovyev, A.; Dutreix, M.; Barillot, E.; Vert, J.-P. Classification of microarray data using gene networks. BMC Bioinform. 2007, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shen, X.; Pan, W. Network-based support vector machine for classification of microarray samples. BMC Bioinform. 2009, 10, S21. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.Y.; Lee, E.; Liu, Y.T.; Lee, D.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xuan, J.; Riggins, R.B.; Wang, Y.; Clarke, R. Identifying protein interaction subnetworks by a bagging Markov random field-based method. Nucleic Acids Res. 2013, 41, e42. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Garrett Injac, S.; Cui, K.; Braun, F.; Lin, Q.; Du, Y.; Zhang, H.; Kogiso, M.; Lindsay, H.; Zhao, S.; et al. Systems biology–based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Sci. Transl. Med. 2018, 10, eaat0150. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, X.; Qi, J.; Yang, C.; Cheng, H.; Ren, Y.; Huang, L. Eight proteins play critical roles in RCC with bone metastasis via mitochondrial dysfunction. Clin. Exp. Metastasis 2015, 32, 605–622. [Google Scholar] [CrossRef]
- Gönen, M.; Margolin, A.A. Drug susceptibility prediction against a panel of drugs using kernelized Bayesian multitask learning. Bioinformatics 2014, 30, i556–i563. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Lobato, D.; Hernández-Lobato, J.M. Bayes Machines for binary classification. Pattern Recognit. Lett. 2008, 29, 1466–1473. [Google Scholar] [CrossRef]
- Miguel Hernández-Lobato, J.; Hernández-Lobato, D.; Suárez, A. Network-based sparse Bayesian classification. Pattern Recognit. 2011, 44, 886–900. [Google Scholar] [CrossRef]
- Yang, J.; Li, A.; Li, Y.; Guo, X.; Wang, M. A novel approach for drug response prediction in cancer cell lines via network representation learning. Bioinformatics 2018, 35, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Brunell, D.; Stephan, C.; Mancuso, J.; Yu, X.; He, B.; Thompson, T.C.; Zinner, R.; Kim, J.; Davies, P.; et al. Driver Network as a Biomarker: Systematic integration and network modeling of multi-omics data to derive driver signaling pathways for drug combination prediction. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, F.; Sheng, J.; Xia, X.; Ma, J.; Zhan, M.; Wong, S.T.C. DrugComboRanker: Drug combination discovery based on target network analysis. Bioinformatics 2014, 30, i228–i236. [Google Scholar] [CrossRef] [PubMed]
- Herbich, R.; Graepel, T.; Campbell, C. Bayes point machines. J. Mach. Learn. Res. 2001, 1, 245–279. [Google Scholar] [CrossRef]
- Minka, T.P. Expectation propagation for approximate Bayesian inference. In Proceedings of the Seventeenth conference on Uncertainty in artificial intelligence, Seattle, WA, USA, 2–5 August 2001; pp. 362–369. [Google Scholar]
- Fröhlich, H.; Sültmann, H.; Brase, J.C.; Johannes, M.; Fälth, M.; Gehrmann, M.; Gade, S.; Beißbarth, T. Integration of pathway knowledge into a reweighted recursive feature elimination approach for risk stratification of cancer patients. Bioinformatics 2010, 26, 2136–2144. [Google Scholar] [CrossRef]
- Symmans, W.F.; Hatzis, C.; Sotiriou, C.; Andre, F.; Peintinger, F.; Regitnig, P.; Daxenbichler, G.; Desmedt, C.; Domont, J.; Marth, C.; et al. Genomic Index of Sensitivity to Endocrine Therapy for Breast Cancer. J. Clin. Oncol. 2010, 28, 4111–4119. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570. [Google Scholar] [CrossRef]
- Benes, C.; Haber, D.A.; Beare, D.; Edelman, E.J.; Lightfoot, H.; Thompson, I.R.; Smith, J.A.; Soares, J.; Stratton, M.R.; Bindal, N.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Awan, A.; Bari, H.; Yan, F.; Moksong, S.; Yang, S.; Chowdhury, S.; Cui, Q.; Yu, Z.; Purisima, E.O.; Wang, E. Regulatory network motifs and hotspots of cancer genes in a mammalian cellular signalling network. IET Syst. Biol. 2007, 1, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Ma, Y.; Jaramillo, M.; Bari, H.; Awan, A.; Yang, S.; Zhang, S.; Liu, L.; Lu, M.; O’Connor-McCourt, M.; et al. A map of human cancer signaling. Mol. Syst. Biol. 2007, 3, 152. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tibiche, C.; Fu, C.; Kaneko, T.; Moran, M.F.; Schiller, M.R.; Li, S.S.; Wang, E. The human phosphotyrosine signaling network: Evolution and hotspots of hijacking in cancer. Genome Res. 2012, 22, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.H.; Hu, J.; Rho, H.S.; Xie, Z.; Woodard, C.; Neiswinger, J.; Cooper, C.; Shirley, M.; Clark, H.M.; Hu, S.; et al. Construction of human activity-based phosphorylation networks. Mol. Syst. Biol. 2013, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. BioCarta. Biotech. Softw. Internet Rep. Comput. Softw. J. Sci. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Muglia, L.J.; Huang, L.F. Network as a Biomarker: A Novel Network-Based Sparse Bayesian Machine for Pathway-Driven Drug Response Prediction. Genes 2019, 10, 602. https://doi.org/10.3390/genes10080602
Liu Q, Muglia LJ, Huang LF. Network as a Biomarker: A Novel Network-Based Sparse Bayesian Machine for Pathway-Driven Drug Response Prediction. Genes. 2019; 10(8):602. https://doi.org/10.3390/genes10080602
Chicago/Turabian StyleLiu, Qi, Louis J. Muglia, and Lei Frank Huang. 2019. "Network as a Biomarker: A Novel Network-Based Sparse Bayesian Machine for Pathway-Driven Drug Response Prediction" Genes 10, no. 8: 602. https://doi.org/10.3390/genes10080602
APA StyleLiu, Q., Muglia, L. J., & Huang, L. F. (2019). Network as a Biomarker: A Novel Network-Based Sparse Bayesian Machine for Pathway-Driven Drug Response Prediction. Genes, 10(8), 602. https://doi.org/10.3390/genes10080602