Changes in the Microbial Community Diversity of Oil Exploitation

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. 16S rRNA Sequencing
2.3. Data Analysis
3. Results and Discussion
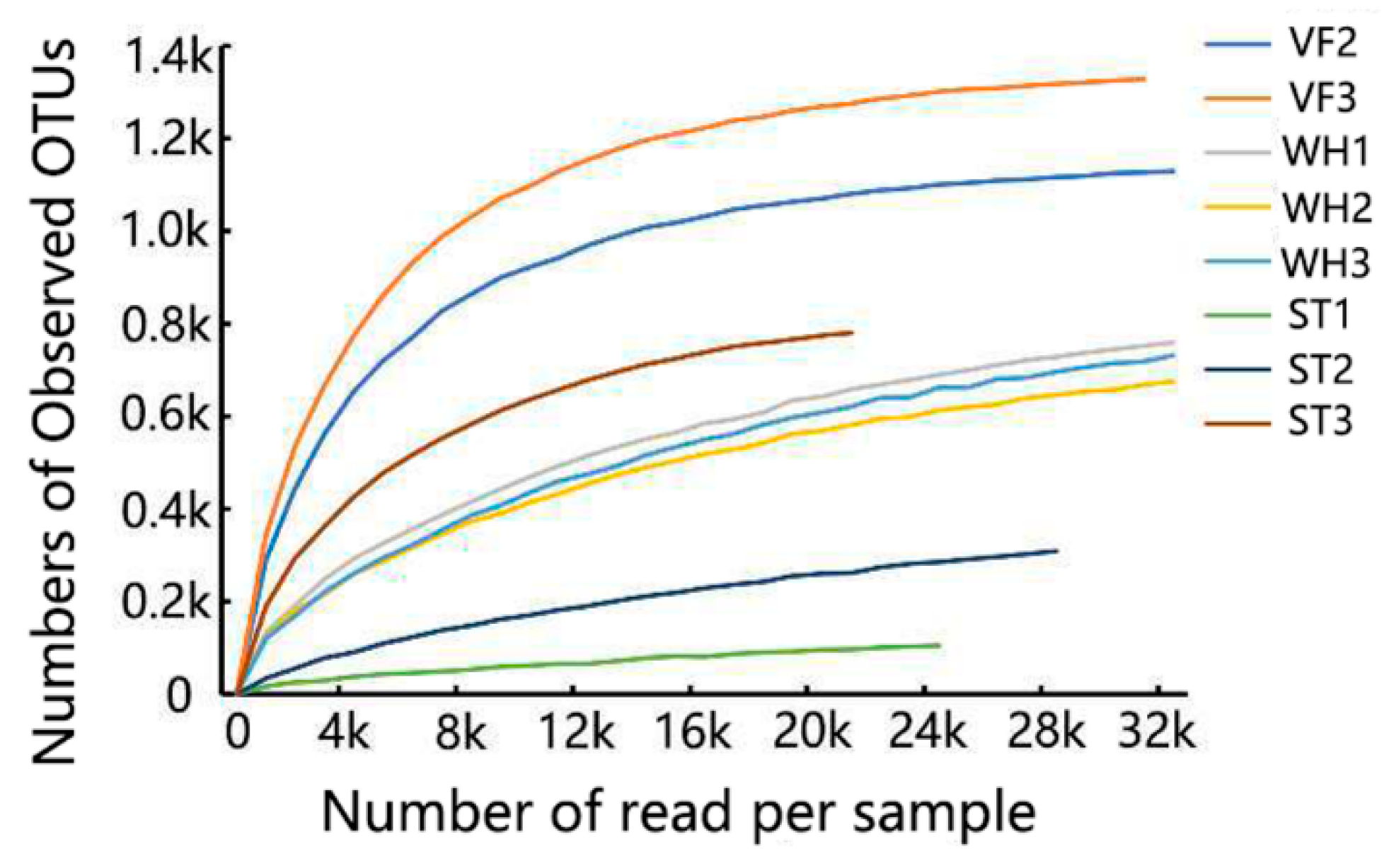
3.1. Microbial Community Abundance
3.2. Microbial Community Diversity Analysis at Phylum Level
3.3. Microbial Community Diversity Analysis at the Genus Level
3.4. Assessment of Microbial Community Function
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| rRNA | ribosomal ribonucleic acid |
| MEOR | microbial enhanced oil recovery technology |
| SRB | sulfate-reducing bacteria |
| MIC | microbiologically influenced corrosion |
| NRB | nitrate and nitrate reducing bacteria |
| FB | fermentative bacteria |
| VF | virgin field |
| WH | wellhead |
| ST | storage tank |
| OUT | operational taxonomic unit |
| PCR | polymerase chain reaction |
| FLASH | fast length adjustment of SHort reads |
References
- Xie, S.; Yang, H.; Luo, G.; Huang, X.; Liu, D.; Wang, Y.; Gong, Y.; Xu, R. Geomicrobial functional groups: A window on the interaction between life and environments. Chin. Sci. Bull. 2012, 57, 2–19. [Google Scholar]
- Castro, H.F.; Classen, A.T.; Austin, E.E.; Norby, R.J.; Schadt, C.W. Soil microbial community responses to multiple experimental climate change drivers. Appl. Environ. Microbiol. 2010, 76, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Rinnan, R.; Michelsen, A.; Bååth, E.; Jonasson, S. Fifteen years of climate change manipulations alter soil microbial communities in a subarctic heath ecosystem. Glob. Chang. Biol. 2010, 13, 28–39. [Google Scholar] [CrossRef]
- Grassia, G.S.; McLean, K.M.; Glénat, P.; Bauld, J.; Sheehy, A.J. A systematic survey for thermophilic fermentative bacteria and archaea in high temperature petroleum reservoirs. FEMS Microbiol. Ecol. 1996, 21, 47–58. [Google Scholar]
- Lazar, I.; Petrisor, I.G.; Yen, T.F. Microbial Enhanced Oil Recovery (MEOR). Pet. Sci. Technol. 2007, 25, 1353–1366. [Google Scholar]
- Zhang, P.; Xu, D.; Li, Y.; Yang, K.; Gu, T. Electron mediators accelerate the microbiologically influenced corrosion of 304 stainless steel by the Desulfovibrio vulgaris biofilm. Bioelectrochemistry 2015, 101, 14–21. [Google Scholar] [PubMed]
- Tian, H.; Gao, P.; Chen, Z.; Li, Y.; Li, Y.; Wang, Y.; Zhou, J.; Li, G.; Ma, T. Compositions and Abundances of Sulfate-Reducing and Sulfur-Oxidizing Microorganisms in Water-Flooded Petroleum Reservoirs with Different Temperatures in China. Front. Microbiol. 2017, 8, 143. [Google Scholar]
- Hubert, C.; Voordouw, G. Oil Field Souring Control by Nitrate-Reducing Sulfurospirillum spp. That Outcompete Sulfate-Reducing Bacteria for Organic Electron Donors. Appl. Environ. Microbiol. 2007, 73, 2644–2652. [Google Scholar]
- Sand, W.; Gehrke, T. Microbially Influenced Corrosion of Steel in Aqueous Environments. Rev. Environ. Sci. Biotechnol. 2003, 2, 169–176. [Google Scholar]
- Bachmann, R.T.; Johnson, A.C.; Edyvean, R.G. Biotechnology in the petroleum industry: An overview. Int. Biodeterior. Biodegrad. 2014, 86, 225–237. [Google Scholar]
- Rechner, A. Colonic metabolism of dietary polyphenols: Influence of structure on microbial fermentation products. Free Radic. Biol. Med. 2004, 36, 212–225. [Google Scholar] [PubMed]
- Yan, L.; Tanaka, S. Ethanol fermentation from biomass resources: Current state and prospects. Appl. Microbiol. Biotechnol. 2006, 69, 627–642. [Google Scholar] [CrossRef]
- Singh, A.; Singh, B.; Ward, O. Potential applications of bioprocess technology in petroleum industry. Biodegradation 2012, 23, 865–880. [Google Scholar] [PubMed]
- Premuzic, E.T.; Lin, M.S. Induced biochemical conversions of heavy crude oils. J. Pet. Sci. Eng. 1999, 22, 171–180. [Google Scholar]
- Watanabe, K.; Kodama, Y.; Syutsubo, K.; Harayama, S. Molecular Characterization of Bacterial Populations in Petroleum-Contaminated Groundwater Discharged from Underground Crude Oil Storage Cavities. Appl. Environ. Microbiol. 2000, 66, 4803–4809. [Google Scholar] [PubMed]
- Van Der Kraan, G.M.; Bruining, J.; Lomans, B.P.; Van Loosdrecht, M.C.M.; Muyzer, G. Microbial diversity of an oil-water processing site and its associated oil field: The possible role of microorganisms as information carriers from oil-associated environments. FEMS Microbiol. Ecol. 2010, 71, 428–443. [Google Scholar] [PubMed]
- Ohtagaki, H.; Yamane, K.; Hattori, Y.; Fujiwara, K. Microbial diversity with dominance of 16S rRNA gene sequences with high GC contents at 74 and 98 °C subsurface crude oil deposits in Japan. FEMS Microbiol. Ecol. 2011, 76, 220–235. [Google Scholar]
- Huang, X.F.; Liu, J.; Lu, L.J.; Wen, Y.; Xu, J.C.; Yang, D.H.; Zhou, Q. Evaluation of screening methods for demulsifying bacteria and characterization of lipopeptide bio-demulsifier produced by Alcaligenes sp. Bioresour. Technol. 2009, 100, 1358–1365. [Google Scholar] [PubMed]
- BIG Data Center Members. Database Resources of the BIG Data Center in 2019. Nucleic Acids Res. 2018, 47, D8–D14. [Google Scholar] [CrossRef]
- Nazina, T.N.; Shestakova, N.M.; Grigor’yan, A.A.; Mikhailova, E.M.; Tourova, T.P.; Poltaraus, A.B.; Feng, C.X.; Ni, F.T.; Belyaev, S.S. Phylogenetic diversity and activity of anaerobic microorganisms of high-temperature horizons of the Dagang oil field (P. R. China). Mikrobiologiia 2006, 75, 55–65. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [PubMed]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to Analyze 16S rRNA Gene Sequences from Microbial Communities. Curr. Protoc. Microbiol. 2012. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Crist, T.O.; Veech, J.A. Additive partitioning of rarefaction curves and species–area relationships: Unifying α-, β- and γ-diversity with sample size and habitat area. Ecol. Lett. 2006, 9, 923–932. [Google Scholar] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar]
- Glaubitz, S.; Abraham, W.R.; Jost, G.; Labrenz, M.; Jürgens, K. Pyruvate utilization by a chemolithoautotrophic epsilonproteobacterial key player of pelagic Baltic Sea redoxclines. FEMS Microbiol. Ecol. 2014, 87, 770–779. [Google Scholar] [CrossRef]
- Cupit, C.; Lomstein, B.A.; Kjeldsen, K.U. Contrasting community composition of endospores and vegetative Firmicutes in a marine sediment suggests both endogenous and exogenous sources of endospore accumulation. Environ. Microbiol. Rep. 2018, 11, 352–360. [Google Scholar]
- Hiraishi, A.; Kishimoto, N.; Kosako, Y.; Wakao, N.; Tano, T. Phylogenetic position of the menaquinone-containing acidophilic chemo-organotroph Acidobacterium capsulatum. FEMS Microbiol. Lett. 2010, 132, 91–94. [Google Scholar] [CrossRef]
- Lin, J.; Hao, B.; Cao, G.; Wang, J.; Feng, Y.; Tan, X.; Wang, W. A study on the microbial community structure in oil reservoirs developed by water flooding. J. Pet. Sci. Eng. 2014, 122, 354–359. [Google Scholar]
- Liu, J.; Fu, B.; Yang, H.; Zhao, M.; He, B.; Zhang, X.H. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: The potential impact of hypoxia and nutrients. Front. Microbiol. 2015, 6, 64. [Google Scholar] [PubMed]
- Abdel-Mawgoud, A.M.; Lépine, F.; Déziel, E. Rhamnolipids: Diversity of structures, microbial origins and roles. Appl. Microbiol. Biotechnol. 2010, 86, 1323–1336. [Google Scholar] [PubMed]
- Satpute, S.K.; Banat, I.M.; Dhakephalkar, P.K.; Banpurkar, A.G.; Chopade, B.A. Biosurfactants, bioemulsifiers and exopolysaccharides from marine microorganisms. Biotechnol. Adv. 2010, 28, 436–450. [Google Scholar] [PubMed]
- Saravanakumari, P.; Mani, K. Structural characterization of a novel xylolipid biosurfactant from Lactococcus lactis and analysis of antibacterial activity against multi-drug resistant pathogens. Bioresour. Technol. 2010, 101, 8851–8854. [Google Scholar] [PubMed]
- Zhang, Z.; Hou, Z.; Yang, C.; Ma, C.; Tao, F.; Xu, P. Degradation of n-alkanes and polycyclic aromatic hydrocarbons in petroleum by a newly isolated Pseudomonas aeruginosa DQ8. Bioresour. Technol. 2011, 102, 4111–4116. [Google Scholar] [PubMed]
- Mogollón, L.; Rodriguez, R.; Larrota, W.; Ortiz, C.; Torres, R. Biocatalytic removal of nickel and vanadium from petroporphyrins and asphaltenes. Biotechnol. Fuels Chem. 1998, 70–72, 765–777. [Google Scholar]
- Rahman, K.; Thahira-Rahman, J.; Lakshmanaperumalsamy, P.; Banat, I.; Rahman, P.; Banat, I. Towards efficient crude oil degradation by a mixed bacterial consortium. Bioresour. Technol. 2002, 85, 257–261. [Google Scholar]
- Zoetendal, E.G.; Collier, C.T.; Koike, S.; Mackie, R.I.; Gaskins, H.R. Molecular ecological analysis of the gastrointestinal microbiota: A Review. J. Nutr. 2004, 134, 465–472. [Google Scholar]
- Reed, S.C.; Cleveland, C.C.; Townsend, A.R. Functional ecology of free-living nitrogen fixation: A contemporary perspective. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 489–512. [Google Scholar] [CrossRef]
- Brewer, T.E.; Handley, K.M.; Carini, P.; Gilbert, J.A.; Fierer, N. Genome reduction in an abundant and ubiquitous soil bacterium ‘Candidatus Udaeobacter copiosus’. Nat. Microbiol. 2017, 2, 16198. [Google Scholar] [CrossRef]
- Hultman, J.; Rahkila, R.; Ali, J.; Rousu, J.; Björkroth, K.J. Meat Processing Plant Microbiome and Contamination Patterns of Cold-Tolerant Bacteria Causing Food Safety and Spoilage Risks in the Manufacture of Vacuum-Packaged Cooked Sausages. Appl. Environ. Microbiol. 2015, 81, 7088–7097. [Google Scholar] [PubMed]
- Roy, A.S.; Baruah, R.; Borah, M.; Singh, A.K.; Boruah, H.P.D.; Saikia, N.; Deka, M.; Dutta, N.; Bora, T.C. Bioremediation potential of native hydrocarbon degrading bacterial strains in crude oil contaminated soil under microcosm study. Int. Biodeterior. Biodegrad. 2014, 94, 79–89. [Google Scholar]
- De León-Lorenzana, A.S.; Delgado-Balbuena, L.; Domínguez-Mendoza, C.A.; Navarro-Noya, Y.E.; Luna-Guido, M.; Dendooven, L. Soil Salinity Controls Relative Abundance of Specific Bacterial Groups Involved in the Decomposition of Maize Plant Residues. Front. Ecol. Evol. 2018, 6, 51. [Google Scholar]
- Childers, M.C.; Daggett, V. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2017, 2, 9–33. [Google Scholar] [PubMed]
- Lauffenburger, D.; Aris, R.; Keller, K. Effects of cell motility and chemotaxis on microbial population growth. Biophys. J. 1982, 40, 209–219. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Number of Reads | Observed OTUs |
|---|---|---|
| VF2 | 33,817 | 1133 |
| VF3 | 31,517 | 1328 |
| WH1 | 33,007 | 764 |
| WH2 | 33,743 | 684 |
| WH3 | 39,734 | 774 |
| ST1 | 24,608 | 107 |
| ST2 | 28,311 | 309 |
| ST3 | 22,769 | 784 |
| Unique genera | Unique Genera in VF | Unique Genera in WH | ||||||
| Caldilinea | Brevibacillus | |||||||
| Coprobacillus | Brochothrix | |||||||
| Nitrosovibrio | Butyrivibrio | |||||||
| Parachlamydia | Capnocytophaga | |||||||
| Sutterella | Bradyrhizobium | |||||||
| Syntrophobacter | Candidatus Rhabdochlamydia | |||||||
| Veillonella | Chryseobacterium | |||||||
| Acidovorax | GOUTA19 | |||||||
| Allobaculum | Hydrogenophilus | |||||||
| Faecalibacterium | Nevskia | |||||||
| Leptotrichia | ||||||||
| Turicibacter | ||||||||
| YRC22 | ||||||||
| Differentially abundant genera | Name | Type | Fold Change | p-value | Name | Type 1 | Fold Change | p-value |
| DA101 | VF/WH | 87 | 5.22 × 10−5 | Lactobacillus | VF/ST | 81.5 | 4.84 × 10−3 | |
| Thermus | VF/WH | 68.2 | 4.89 × 10−4 | Sphingobacterium | VF/ST | 81 | 7.25 × 10−5 | |
| Anaeromyxobacter | VF/WH | 35.3 | 1.88 × 10−3 | Oscillospira | VF/ST | 59.6 | 8.38 × 10−4 | |
| Methylibium | VF/WH | 34.9 | 9.80 × 10−4 | Stenotrophomonas | VF/ST | 53.4 | 1.19 × 10−4 | |
| Adlercreutzia | VF/WH | 20.4 | 1.58 × 10−3 | Devosia | VF/ST | 45.8 | 9.82 × 10−3 | |
| Exiguobacterium | VF/WH | 16.5 | 2.59 × 10−3 | Streptomyces | VF/ST | 34.5 | 1.60 × 10−3 | |
| Deinococcus | VF/WH | 15.7 | 9.48 × 10−3 | Akkermansia | VF/ST | 31.5 | 1.00 × 10−2 | |
| Nitrospira | VF/WH | 14.8 | 1.56 × 10−4 | Methylobacterium | VF/ST | 24.5 | 1.87 × 10−3 | |
| Oscillospira | VF/WH | 14 | 8.31 × 10−4 | Planomicrobium | VF/ST | 21 | 3.15 × 10−3 | |
| Psychrobacter | VF/WH | 13.5 | 1.25 × 10−3 | Acinetobacter | VF/ST | 19.3 | 4.10 × 10−4 | |
| Desulfovibrio | VF/WH | 10.5 | 8.54 × 10−3 | Brevibacterium | VF/ST | 17.3 | 6.32 × 10−3 | |
| Clostridium | VF/WH | 7.5 | 8.28 × 10−4 | Prauseria | VF/ST | 1.15 × 10−4 | 2.13 × 10−3 | |
| Ochrobactrum | VF/WH | 7.1 | 3.11 × 10−3 | Burkholderia | WH/ST | 3708 | 6.76 × 10−4 | |
| Rhodanobacter | VF/WH | 5.4 | 1.28 × 10−3 | Sediminibacterium | WH/ST | 650.1 | 9.29 × 10−4 | |
| Staphylococcus | VF/WH | 4.5 | 5.12 × 10−3 | Ralstonia | WH/ST | 154.6 | 3.44 × 10−4 | |
| Stenotrophomonas | VF/WH | 4.5 | 8.16 × 10−3 | Carnobacterium | WH/ST | 123.9 | 5.18 × 10−5 | |
| Acinetobacter | VF/WH | 4.1 | 1.07 × 10−3 | Geobacillus | WH/ST | 109.2 | 2.11 × 10−4 | |
| Bacteroides | VF/WH | 3.9 | 3.22 × 10−3 | Lactococcus | WH/ST | 89.3 | 8.00 × 10−5 | |
| Microbacterium | VF/WH | 3.3 | 5.81 × 10−3 | Fusobacterium | WH/ST | 82 | 5.36 × 10−3 | |
| Anoxybacillus | VF/WH | 0.2 | 3.45 × 10−4 | Streptococcus | WH/ST | 81.4 | 1.98 × 10−4 | |
| Streptococcus | VF/WH | 0.1 | 2.79 × 10−3 | Leuconostoc | WH/ST | 65.8 | 2.59 × 10−3 | |
| Lactococcus | VF/WH | 3.09 × 10−2 | 1.09 × 10−3 | Staphylococcus | WH/ST | 51 | 1.33 × 10−3 | |
| Geobacillus | VF/WH | 1.12 × 10−2 | 2.13 × 10−3 | Methylobacterium | WH/ST | 35 | 4.15 × 10−3 | |
| Carnobacterium | VF/WH | 1.73 × 10−3 | 7.21 × 10−4 | Janthinobacterium | WH/ST | 32.5 | 1.59 × 10−4 | |
| Burkholderia | VF/ST | 2068.5 | 7.80 × 10−3 | Ruminococcus | WH/ST | 27.5 | 1.43 × 10−4 | |
| Thermus | VF/ST | 386.5 | 4.20 × 10−4 | Arthrobacter | WH/ST | 23.5 | 4.92 × 10−4 | |
| Ralstonia | VF/ST | 372.7 | 7.16 × 10−3 | Trueperella | WH/ST | 22 | 7.14 × 10−3 | |
| Deinococcus | VF/ST | 235.5 | 7.15 × 10−3 | Lactobacillus | WH/ST | 20.5 | 1.83 × 10−3 | |
| Staphylococcus | VF/ST | 231 | 2.35 × 10−3 | Ruminococcus | WH/ST | 18 | 1.00 × 10−2 | |
| Rhodococcus | VF/ST | 141 | 7.17 × 10−3 | Lysobacter | WH/ST | 17 | 7.60 × 10−4 | |
| Enhydrobacter | VF/ST | 114.8 | 7.95 × 10−3 | Pseudomonas | WH/ST | 15 | 4.74 × 10−3 | |
| Ochrobactrum | VF/ST | 100 | 1.30 × 10−3 | Anoxybacillus | WH/ST | 10.1 | 1.41 × 10−3 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wu, J.; Lin, J.; Zhao, J.; Xu, T.; Yang, Q.; Zhao, J.; Zhao, Z.; Song, X. Changes in the Microbial Community Diversity of Oil Exploitation. Genes 2019, 10, 556. https://doi.org/10.3390/genes10080556
Liu J, Wu J, Lin J, Zhao J, Xu T, Yang Q, Zhao J, Zhao Z, Song X. Changes in the Microbial Community Diversity of Oil Exploitation. Genes. 2019; 10(8):556. https://doi.org/10.3390/genes10080556
Chicago/Turabian StyleLiu, Jingjing, Jing Wu, Jiawei Lin, Jian Zhao, Tianyi Xu, Qichang Yang, Jing Zhao, Zhongming Zhao, and Xiaofeng Song. 2019. "Changes in the Microbial Community Diversity of Oil Exploitation" Genes 10, no. 8: 556. https://doi.org/10.3390/genes10080556
APA StyleLiu, J., Wu, J., Lin, J., Zhao, J., Xu, T., Yang, Q., Zhao, J., Zhao, Z., & Song, X. (2019). Changes in the Microbial Community Diversity of Oil Exploitation. Genes, 10(8), 556. https://doi.org/10.3390/genes10080556