Preserving Genome Integrity during the Early Embryonic DNA Replication Cycles


Abstract
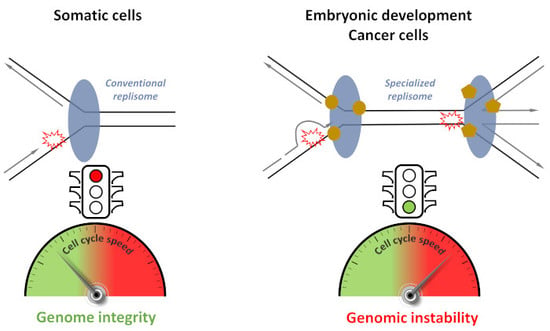
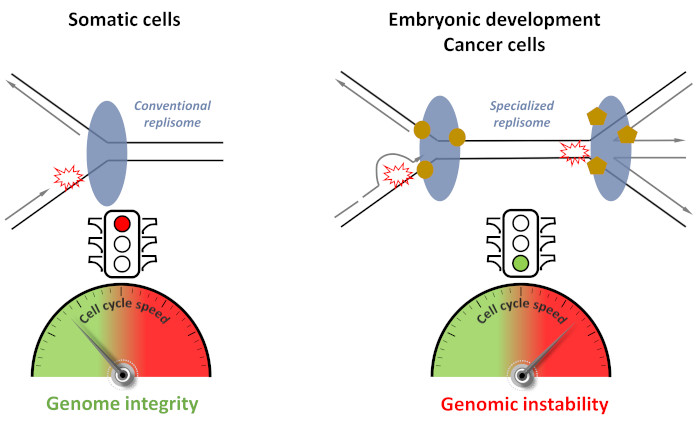
1. Introduction
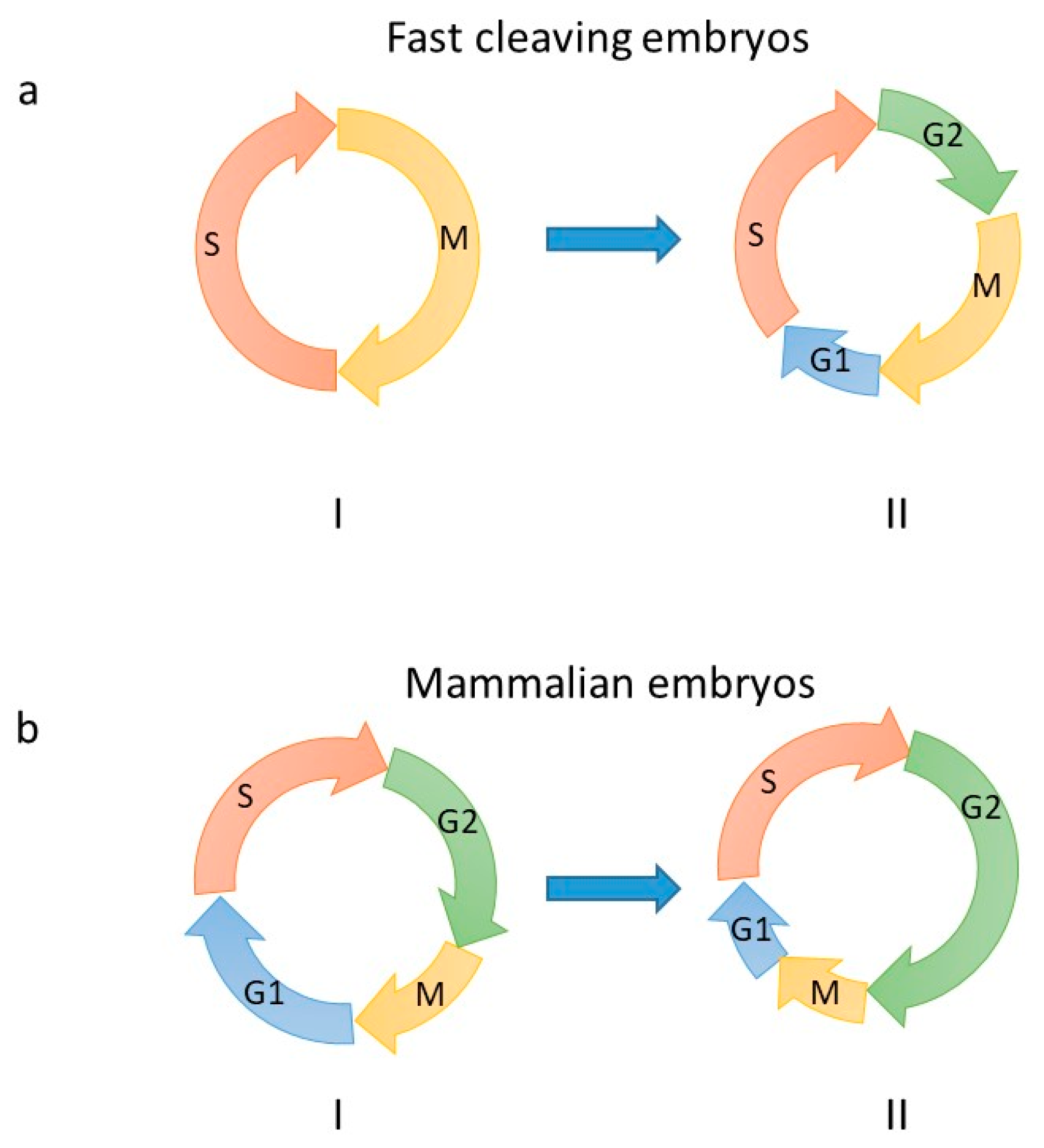
2. Embryonic DNA Replication
3. Preserving Genome Integrity during Early Embryogenesis Replication Cycles
3.1. G1/S Checkpoint
3.2. S-Phase Checkpoint
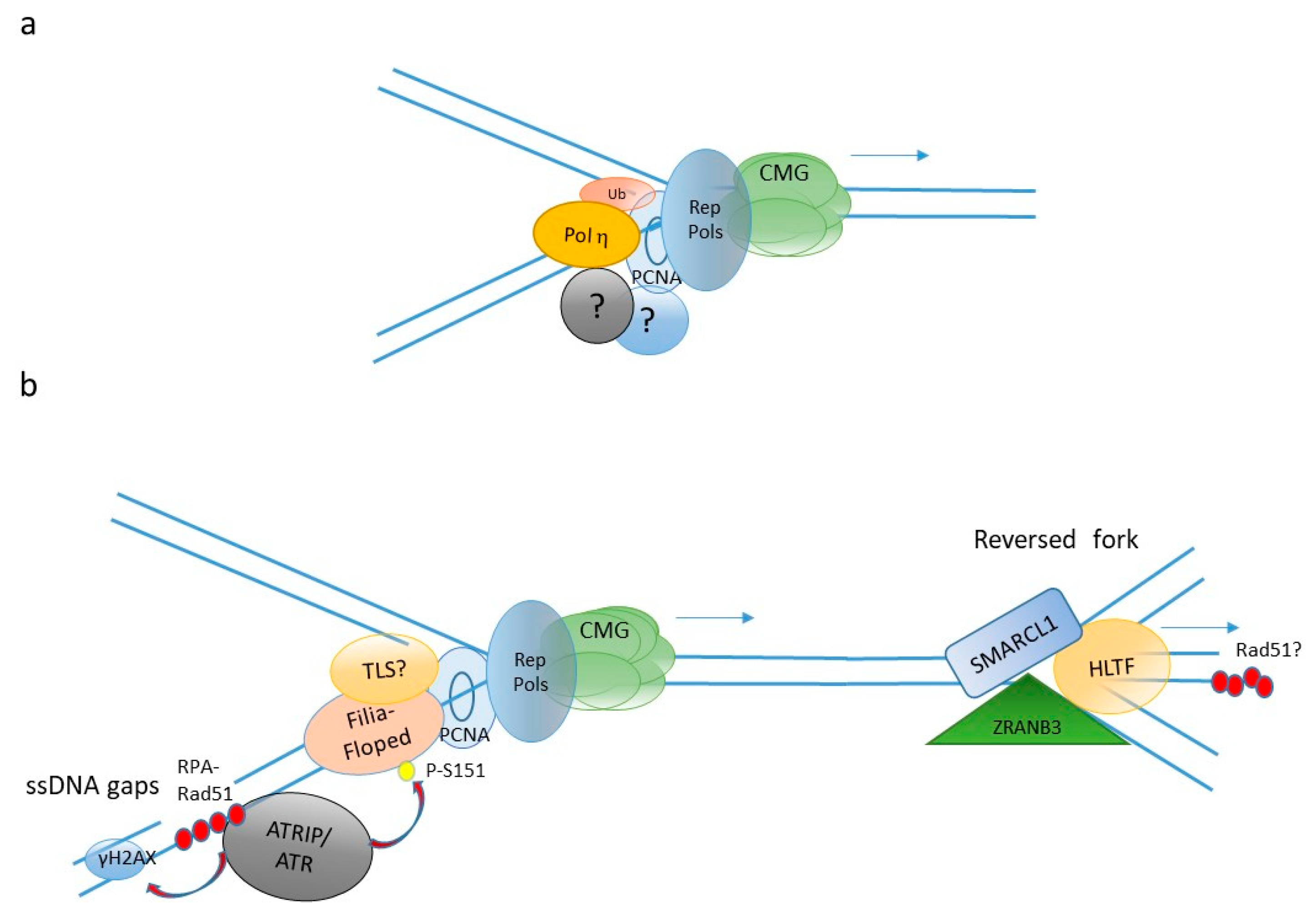
4. DNA Repair and DNA Damage Tolerance in the Embryo
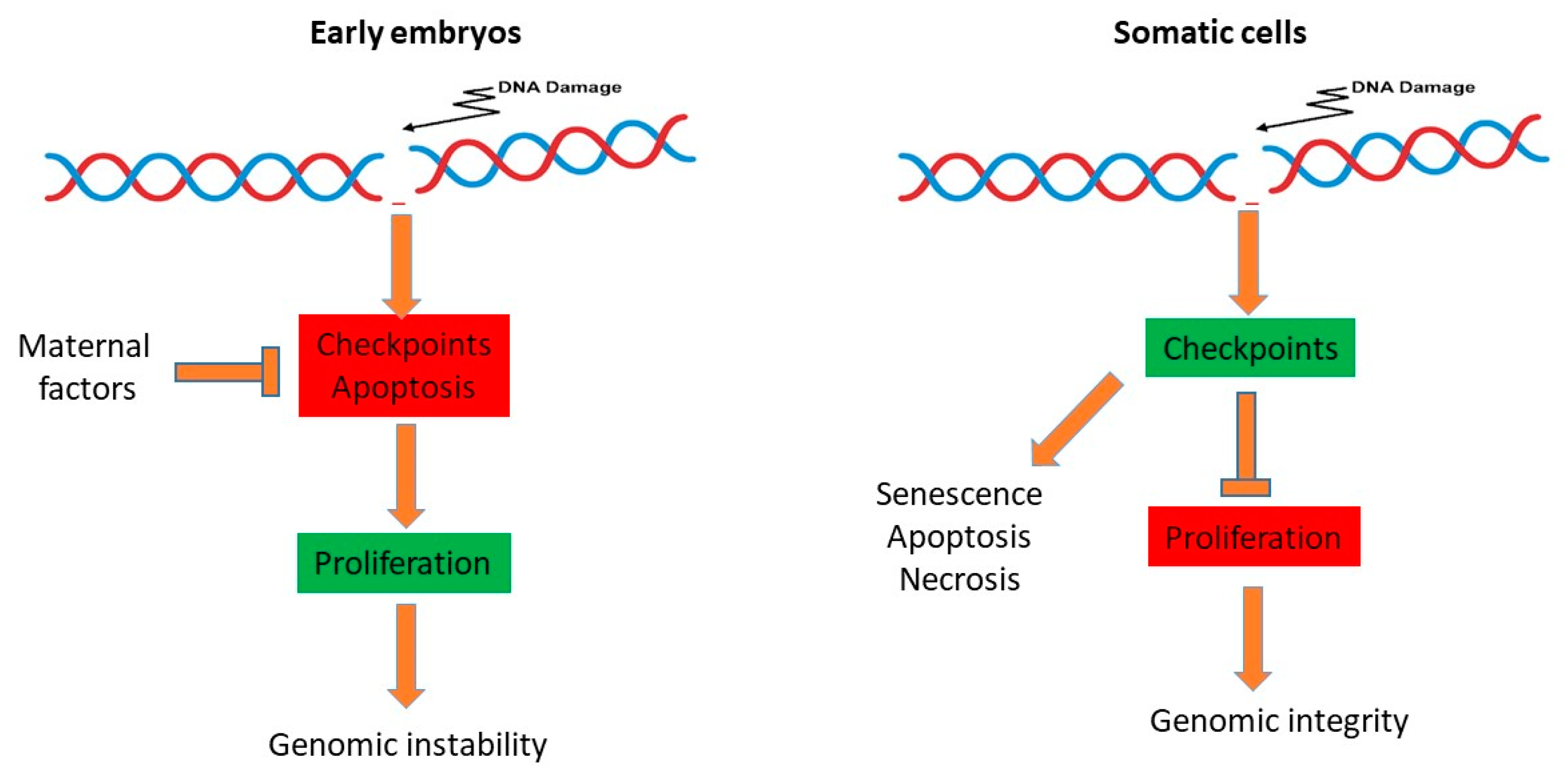
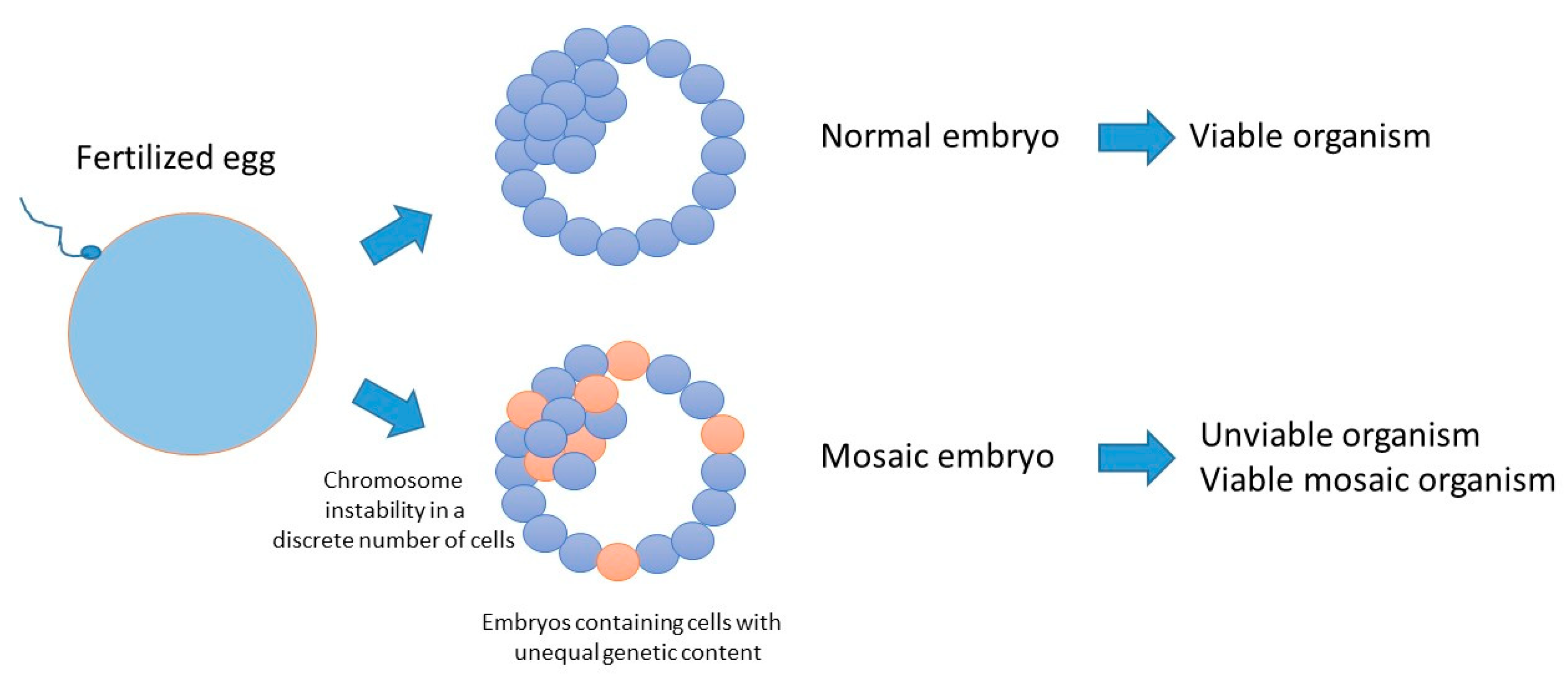
5. Consequences of Reduced Genome Surveillance in the Early Embryo
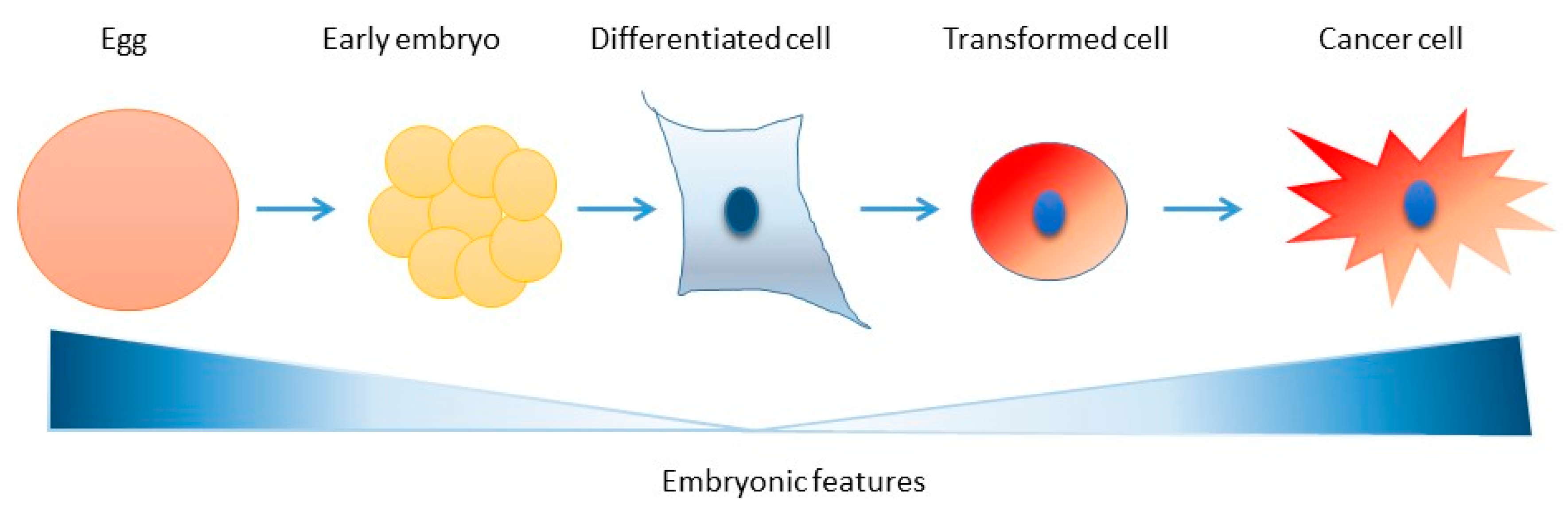
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hakem, R. DNA-Damage Repair, the Good, the Bad, and the Ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, A.J.; Weinberg, C.R.; O’Connor, J.F.; Baird, D.D.; Schlatterer, J.P.; Canfield, R.E.; Armstrong, E.G.; Nisula, B.C. Incidence of Early Loss of Pregnancy. N. Engl. J. Med. 1988, 319, 189–194. [Google Scholar] [CrossRef]
- Warkentin, K.M. Adaptive Plasticity in Hatching Age: A Response to Predation Risk Trade-Offs. Proc. Natl. Acad. Sci. USA 1995, 92, 3507–3510. [Google Scholar] [CrossRef] [PubMed]
- Prioleau, M.N.; Huet, J.; Sentenac, A.; Mechali, M. Competition between Chromatin and Transcription Complex Assembly Regulates Gene Expression during Early Development. Cell 1994, 77, 439–449. [Google Scholar] [CrossRef]
- Amodeo, A.A.; Jukam, D.; Straight, A.F.; Skotheim, J.M. Histone Titration against the Genome Sets the DNA-to-Cytoplasm Threshold for the Xenopus Midblastula Transition. Proc. Natl. Acad. Sci. USA 2015, 112, E1086–E1095. [Google Scholar] [CrossRef]
- Almouzni, G.; Wolffe, A.P. Constraints on Transcriptional Activator Function Contribute to Transcriptional Quiescence during Early Xenopus Embryogenesis. EMBO J. 1995, 14, 1752–1765. [Google Scholar] [CrossRef]
- Lemaitre, J.M.; Geraud, G.; Mechali, M. Dynamics of the Genome during Early Xenopus Laevis Development: Karyomeres as Independent Units of Replication. J. Cell Biol. 1998, 142, 1159–1166. [Google Scholar] [CrossRef]
- Sasaki, T.; Sawado, T.; Yamaguchi, M.; Shinomiya, T. Specification of Regions of DNA Replication Initiation during Embryogenesis in the 65-Kilobase DNApolalpha-DE2F Locus of Drosophila Melanogaster. Mol. Cell. Biol. 1999, 19, 547–555. [Google Scholar] [CrossRef]
- Hyrien, O.; Maric, C.; Mechali, M. Transition in Specification of Embryonic Metazoan DNA Replication Origins. Science 1995, 270, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Danis, E.; Brodolin, K.; Menut, S.; Maiorano, D.; Girard-Reydet, C.; Mechali, M. Specification of a DNA Replication Origin by a Transcription Complex. Nat. Cell Biol. 2004, 6, 721–730. [Google Scholar] [CrossRef]
- DePamphilis, M.L. Genome Duplication at the Beginning of Mammalian Development. Curr. Top. Dev. Biol. 2016, 120, 55–102. [Google Scholar] [CrossRef] [PubMed]
- Reusswig, K.-U.; Pfander, B. Control of Eukaryotic DNA Replication Initiation-Mechanisms to Ensure Smooth Transitions. Genes 2019, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Kermi, C.; Prieto, S.; van der Laan, S.; Tsanov, N.; Recolin, B.; Uro-Coste, E.; Delisle, M.B.; Maiorano, D. RAD18 Is a Maternal Limiting Factor Silencing the UV-Dependent DNA Damage Checkpoint in Xenopus Embryos. Dev. Cell 2015, 34, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.; Baxley, R.M.; Moldovan, G.-L.; Bielinsky, A.-K. Mechanisms of DNA Damage Tolerance: Post-Translational Regulation of PCNA. Genes 2018, 10, 10. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harbor Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Tikhmyanova, N.; Coleman, T.R. Isoform Switching of Cdc6 Contributes to Developmental Cell Cycle Remodeling. Dev. Biol. 2003, 260, 362–375. [Google Scholar] [CrossRef]
- Sible, J.C.; Erikson, E.; Hendrickson, M.; Maller, J.L.; Gautier, J. Developmental Regulation of MCM Replication Factors in Xenopus Laevis. Curr. Biol. 1998, 8, 347–350. [Google Scholar] [CrossRef]
- Takahashi, T.S.; Walter, J.C. Cdc7-Drf1 Is a Developmentally Regulated Protein Kinase Required for the Initiation of Vertebrate DNA Replication. Genes Dev. 2005, 19, 2295–2300. [Google Scholar] [CrossRef]
- Silva, T.; Bradley, R.H.; Gao, Y.; Coue, M. Xenopus CDC7/DRF1 Complex Is Required for the Initiation of DNA Replication. J. Biol. Chem. 2006, 281, 11569–11576. [Google Scholar] [CrossRef] [PubMed]
- Collart, C.; Smith, J.C.; Zegerman, P. Chk1 Inhibition of the Replication Factor Drf1 Guarantees Cell-Cycle Elongation at the Xenopus Laevis Mid-Blastula Transition. Dev. Cell 2017, 42, 82–96.e3. [Google Scholar] [CrossRef] [PubMed]
- Christov, C.P.; Gardiner, T.J.; Szüts, D.; Krude, T. Functional Requirement of Noncoding Y RNAs for Human Chromosomal DNA Replication. Mol. Cell. Biol. 2006, 26, 6993–7004. [Google Scholar] [CrossRef] [PubMed]
- Collart, C.; Christov, C.P.; Smith, J.C.; Krude, T. The Midblastula Transition Defines the Onset of Y RNA-Dependent DNA Replication in Xenopus Laevis. Mol. Cell. Biol. 2011, 31, 3857–3870. [Google Scholar] [CrossRef] [PubMed]
- Christov, C.P.; Dingwell, K.S.; Skehel, M.; Wilkes, H.S.; Sale, J.E.; Smith, J.C.; Krude, T. A NuRD Complex from Xenopus Laevis Eggs Is Essential for DNA Replication during Early Embryogenesis. Cell Rep. 2018, 22, 2265–2278. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, W.; Cun, Y.; Li, J.; Liu, Y.; Gao, J.; Zhu, H.; Zhou, H.; Zhang, R.; Zheng, P. Mouse Embryonic Stem Cells Have Increased Capacity for Replication Fork Restart Driven by the Specific Filia-Floped Protein Complex. Cell Res. 2018, 28, 69–89. [Google Scholar] [CrossRef]
- Ahuja, A.K.; Jodkowska, K.; Teloni, F.; Bizard, A.H.; Zellweger, R.; Herrador, R.; Ortega, S.; Hickson, I.D.; Altmeyer, M.; Mendez, J.; et al. A Short G1 Phase Imposes Constitutive Replication Stress and Fork Remodelling in Mouse Embryonic Stem Cells. Nat. Commun. 2016, 7, 10660. [Google Scholar] [CrossRef]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef]
- Banath, J.P.; Banuelos, C.A.; Klokov, D.; MacPhail, S.M.; Lansdorp, P.M.; Olive, P.L. Explanation for Excessive DNA Single-Strand Breaks and Endogenous Repair Foci in Pluripotent Mouse Embryonic Stem Cells. Exp. Cell Res. 2009, 315, 1505–1520. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls That Ensure the Order of Cell Cycle Events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Eychenie, S.; Decaillet, C.; Basbous, J.; Constantinou, A. DNA Structure-Specific Priming of ATR Activation by DNA-PKcs. J. Cell Biol. 2013, 202, 421–429. [Google Scholar] [CrossRef]
- Lin, Y.F.; Shih, H.Y.; Shang, Z.; Matsunaga, S.; Chen, B.P. DNA-PKcs Is Required to Maintain Stability of Chk1 and Claspin for Optimal Replication Stress Response. Nucleic Acids Res. 2014, 42, 4463–4473. [Google Scholar] [CrossRef] [PubMed]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK Phosphorylation of RPA32 Ser4/Ser8 Regulates Replication Stress Checkpoint Activation, Fork Restart, Homologous Recombination and Mitotic Catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Van, C.; Yan, S.; Michael, W.M.; Waga, S.; Cimprich, K.A. Continued Primer Synthesis at Stalled Replication Forks Contributes to Checkpoint Activation. J. Cell Biol. 2010, 189, 233–246. [Google Scholar] [CrossRef]
- Betous, R.; Pillaire, M.J.; Pierini, L.; van der Laan, S.; Recolin, B.; Ohl-Seguy, E.; Guo, C.; Niimi, N.; Gruz, P.; Nohmi, T.; et al. DNA Polymerase Kappa-Dependent DNA Synthesis at Stalled Replication Forks Is Important for CHK1 Activation. EMBO J. 2013, 32, 2172–2185. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.P.; Lindsey-Boltz, L.A.; Cesare, A.J.; Maniwa, Y.; Griffith, J.D.; Hurwitz, J.; Sancar, A. Loading of the Human 9-1-1 Checkpoint Complex onto DNA by the Checkpoint Clamp Loader HRad17-Replication Factor C Complex in Vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 1633–1638. [Google Scholar] [CrossRef]
- Ellison, V.; Stillman, B. Biochemical Characterization of DNA Damage Checkpoint Complexes: Clamp Loader and Clamp Complexes with Specificity for 5′ Recessed DNA. PLoS Biol. 2003, 1, E33. [Google Scholar] [CrossRef]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The Impact of Replication Stress on Replication Dynamics and DNA Damage in Vertebrate Cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Zhou, Q.; Chen, J.; Yuan, J. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr. Biol. 2016, 26, 3257–3268. [Google Scholar] [CrossRef]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 Acts at Stalled Replication Forks to Maintain Genome Integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR Kinase by the RPA-Binding Protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef]
- Feng, S.; Zhao, Y.; Xu, Y.; Ning, S.; Huo, W.; Hou, M.; Gao, G.; Ji, J.; Guo, R.; Xu, D. Ewing Tumor-Associated Antigen 1 Interacts with Replication Protein A to Promote Restart of Stalled Replication Forks. J. Biol. Chem. 2016, 291, 21956–21962. [Google Scholar] [CrossRef]
- Childs, E.J.; Mocci, E.; Campa, D.; Bracci, P.M.; Gallinger, S.; Goggins, M.; Li, D.; Neale, R.E.; Olson, S.H.; Scelo, G.; et al. Common Variation at 2p13.3, 3q29, 7p13 and 17q25.1 Associated with Susceptibility to Pancreatic Cancer. Nat. Genet. 2015, 47, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Recolin, B.; van der Laan, S.; Tsanov, N.; Maiorano, D. Molecular Mechanisms of DNA Replication Checkpoint Activation. Genes 2014, 5, 147–175. [Google Scholar] [CrossRef]
- Adiga, S.K.; Toyoshima, M.; Shiraishi, K.; Shimura, T.; Takeda, J.; Taga, M.; Nagai, H.; Kumar, P.; Niwa, O. P21 Provides Stage Specific DNA Damage Control to Preimplantation Embryos. Oncogene 2007, 26, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Adiga, S.K.; Toyoshima, M.; Shimura, T.; Takeda, J.; Uematsu, N.; Niwa, O. Delayed and Stage Specific Phosphorylation of H2AX during Preimplantation Development of γ-Irradiated Mouse Embryos. Reproduction 2007, 133, 415–422. [Google Scholar] [CrossRef]
- Wang, H.; Luo, Y.; Lin, Z.; Lee, I.-W.; Kwon, J.; Cui, X.-S.; Kim, N.-H. Effect of ATM and HDAC Inhibition on Etoposide-Induced DNA Damage in Porcine Early Preimplantation Embryos. PLoS ONE 2015, 10, e0142561. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, A.A.; Bletsa, R.; Desmarais, B.; Mara, C.; Kallianidis, K.; Loutradis, D. Genome-Wide Microarray Evidence That 8-Cell Human Blastomeres over-Express Cell Cycle Drivers and under-Express Checkpoints. J. Assist. Reprod. Genet. 2010, 27, 265–276. [Google Scholar] [CrossRef]
- Clute, P.; Masui, Y. Microtubule Dependence of Chromosome Cycles in Xenopus Laevis Blastomeres under the Influence of a DNA Synthesis Inhibitor, Aphidicolin. Dev. Biol. 1997, 185, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fasulo, B.; Koyama, C.; Yu, K.R.; Homola, E.M.; Hsieh, T.S.; Campbell, S.D.; Sullivan, W. Chk1 and Wee1 Kinases Coordinate DNA Replication, Chromosome Condensation, and Anaphase Entry. Mol. Biol. Cell 2012, 23, 1047–1057. [Google Scholar] [CrossRef]
- Sibon, O.C.; Laurençon, A.; Hawley, R.; Theurkauf, W.E. The Drosophila ATM Homologue Mei-41 Has an Essential Checkpoint Function at the Midblastula Transition. Curr. Biol. 1999, 9, 302–312. [Google Scholar] [CrossRef]
- Laurençon, A.; Purdy, A.; Sekelsky, J.; Hawley, R.S.; Su, T.T. Phenotypic Analysis of Separation-of-Function Alleles of MEI-41, Drosophila ATM/ATR. Genetics 2003, 164, 589–601. [Google Scholar]
- Fogarty, P.; Campbell, S.D.; Abu-Shumays, R.; Phalle, B.S.; Yu, K.R.; Uy, G.L.; Goldberg, M.L.; Sullivan, W. The Drosophila Grapes Gene Is Related to Checkpoint Gene Chk1/Rad27 and Is Required for Late Syncytial Division Fidelity. Curr. Biol. 1997, 7, 418–426. [Google Scholar] [CrossRef]
- Kalogeropoulos, N.; Christoforou, C.; Green, A.J.; Gill, S.; Ashcroft, N.R. Chk-1 Is an Essential Gene and Is Required for an S-M Checkpoint during Early Embryogenesis. Cell Cycle 2004, 3, 1196–1200. [Google Scholar] [CrossRef][Green Version]
- Sibon, O.C.; Stevenson, V.A.; Theurkauf, W.E. DNA-Replication Checkpoint Control at the Drosophila Midblastula Transition. Nature 1997, 388, 93–97. [Google Scholar] [CrossRef]
- Takada, S.; Kwak, S.; Koppetsch, B.S.; Theurkauf, W.E. Grp (Chk1) Replication-Checkpoint Mutations and DNA Damage Trigger a Chk2-Dependent Block at the Drosophila Midblastula Transition. Development 2007, 134, 1737–1744. [Google Scholar] [CrossRef]
- Price, D.; Rabinovitch, S.; O’Farrell, P.H.; Campbell, S.D. Drosophila Wee1 Has an Essential Role in the Nuclear Divisions of Early Embryogenesis. Genetics 2000, 155, 159–166. [Google Scholar]
- Galli, M.; Morgan, D.O. Cell Size Determines the Strength of the Spindle Assembly Checkpoint during Embryonic Development. Dev. Cell 2016, 36, 344–352. [Google Scholar] [CrossRef]
- Ikegami, R.; Zhang, J.; Rivera-Bennetts, A.K.; Yager, T.D. Activation of the Metaphase Checkpoint and an Apoptosis Programme in the Early Zebrafish Embryo, by Treatment with the Spindle-Destabilising Agent Nocodazole. Zygote 1997, 5, 329–350. [Google Scholar] [CrossRef]
- Minshull, J.; Sun, H.; Tonks, N.K.; Murray, A.W. A MAP Kinase-Dependent Spindle Assembly Checkpoint in Xenopus Egg Extracts. Cell 1994, 79, 475–486. [Google Scholar] [CrossRef]
- Anderson, J.A.; Lewellyn, A.L.; Maller, J.L. Ionizing Radiation Induces Apoptosis and Elevates Cyclin A1-Cdk2 Activity before but Not after the Midblastula Transition in Xenopus. Mol. Biol. Cell 1997, 8, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Hensey, C.; Gautier, J. A Developmental Timer That Regulates Apoptosis at the Onset of Gastrulation. Mech. Dev. 1997, 69, 183–195. [Google Scholar] [CrossRef]
- Bazrgar, M.; Gourabi, H.; Yazdi, P.E.; Vazirinasab, H.; Fakhri, M.; Hassani, F.; Valojerdi, M.R. DNA Repair Signalling Pathway Genes Are Overexpressed in Poor-Quality Pre-Implantation Human Embryos with Complex Aneuploidy. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 175, 152–156. [Google Scholar] [CrossRef]
- Fear, J.M.; Hansen, P.J. Developmental Changes in Expression of Genes Involved in Regulation of Apoptosis in the Bovine Preimplantation Embryo. Biol. Reprod. 2011, 84, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Pauklin, S.; Vallier, L. The Cell-Cycle State of Stem Cells Determines Cell Fate Propensity. Cell 2013, 155, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, S.; Tsanov, N.; Crozet, C.; Maiorano, D. High Dub3 Expression in Mouse ESCs Couples the G1/S Checkpoint to Pluripotency. Mol. Cell 2013, 52, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Jaroudi, S.; SenGupta, S. DNA Repair in Mammalian Embryos. Mutat. Res. 2007, 635, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Langley, A.R.; Smith, J.C.; Stemple, D.L.; Harvey, S.A. New Insights into the Maternal to Zygotic Transition. Development 2014, 141, 3834–3841. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Amariglio, F.; Tchang, F.; Prioleau, M.N.; Soussi, T.; Cibert, C.; Méchali, M. A Functional Analysis of P53 during Early Development of Xenopus Laevis. Oncogene 1997, 15, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Aladjem, M.I.; Spike, B.T.; Rodewald, L.W.; Hope, T.J.; Klemm, M.; Jaenisch, R.; Wahl, G.M. ES Cells Do Not Activate P53-Dependent Stress Responses and Undergo P53-Independent Apoptosis in Response to DNA Damage. Curr. Biol. 1998, 8, 145–155. [Google Scholar] [CrossRef]
- Prost, S.; Bellamy, C.O.; Clarke, A.R.; Wyllie, A.H.; Harrison, D.J. P53-Independent DNA Repair and Cell Cycle Arrest in Embryonic Stem Cells. FEBS Lett. 1998, 425, 499–504. [Google Scholar] [CrossRef]
- Suvorova, I.I.; Grigorash, B.B.; Chuykin, I.A.; Pospelova, T.V.; Pospelov, V.A. G1 Checkpoint Is Compromised in Mouse ESCs Due to Functional Uncoupling of P53-P21Waf1 Signaling. Cell Cycle 2016, 15, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gou, H.; Tripathi, B.K.; Huang, J.; Jiang, S.; Dubois, W.; Waybright, T.; Lei, M.; Shi, J.; Zhou, M.; et al. An Apela RNA-Containing Negative Feedback Loop Regulates P53-Mediated Apoptosis in Embryonic Stem Cells. Cell Stem Cell 2015, 16, 669–683. [Google Scholar] [CrossRef]
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct Regulatory Mechanisms and Functions for P53-Activated and P53-Repressed DNA Damage Response Genes in Embryonic Stem Cells. Mol. Cell 2012, 46, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Solozobova, V.; Rolletschek, A.; Blattner, C. Nuclear Accumulation and Activation of P53 in Embryonic Stem Cells after DNA Damage. BMC Cell Biol. 2009, 10, 46. [Google Scholar] [CrossRef]
- Desmarais, J.A.; Hoffmann, M.J.; Bingham, G.; Gagou, M.E.; Meuth, M.; Andrews, P.W. Human Embryonic Stem Cells Fail to Activate CHK1 and Commit to Apoptosis in Response to DNA Replication Stress. Stem Cells 2012, 30, 1385–1393. [Google Scholar] [CrossRef]
- Barta, T.; Vinarsky, V.; Holubcova, Z.; Dolezalova, D.; Verner, J.; Pospisilova, S.; Dvorak, P.; Hampl, A. Human Embryonic Stem Cells Are Capable of Executing G1/S Checkpoint Activation. Stem Cells 2010, 28, 1143–1152. [Google Scholar] [CrossRef]
- Wang, X.; Liu, D.; He, D.; Suo, S.; Xia, X.; He, X.; Han, J.-D.J.; Zheng, P. Transcriptome Analyses of Rhesus Monkey Preimplantation Embryos Reveal a Reduced Capacity for DNA Double-Strand Break Repair in Primate Oocytes and Early Embryos. Genome Res. 2017, 27, 567–579. [Google Scholar] [CrossRef]
- Raff, J.W.; Glover, D.M. Nuclear and Cytoplasmic Mitotic Cycles Continue in Drosophila Embryos in Which DNA Synthesis Is Inhibited with Aphidicolin. J. Cell Biol. 1988, 107, 2009–2019. [Google Scholar] [CrossRef]
- Holway, A.H.; Kim, S.H.; La Volpe, A.; Michael, W.M. Checkpoint Silencing during the DNA Damage Response in Caenorhabditis Elegans Embryos. J. Cell Biol. 2006, 172, 999–1008. [Google Scholar] [CrossRef]
- Ikegami, R.; Rivera-Bennetts, A.K.; Brooker, D.L.; Yager, T.D. Effect of Inhibitors of DNA Replication on Early Zebrafish Embryos: Evidence for Coordinate Activation of Multiple Intrinsic Cell-Cycle Checkpoints at the Mid-Blastula Transition. Zygote 1997, 5, 153–175. [Google Scholar] [CrossRef]
- Dasso, M.; Newport, J.W. Completion of DNA Replication Is Monitored by a Feedback System That Controls the Initiation of Mitosis in Vitro: Studies in Xenopus. Cell 1990, 61, 811–823. [Google Scholar] [CrossRef]
- Newport, J.; Kirschner, M. A Major Developmental Transition in Early Xenopus Embryos: I. Characterization and Timing of Cellular Changes at the Midblastula Stage. Cell 1982, 30, 675–686. [Google Scholar] [CrossRef]
- Kimelman, D.; Kirschner, M.; Scherson, T. The Events of the Midblastula Transition in Xenopus Are Regulated by Changes in the Cell Cycle. Cell 1987, 48, 399–407. [Google Scholar] [CrossRef]
- Newport, J.; Dasso, M. On the Coupling between DNA Replication and Mitosis. J. Cell Sci. Suppl. 1989, 12, 149–160. [Google Scholar] [CrossRef]
- Kappas, N.C.; Savage, P.; Chen, K.C.; Walls, A.T.; Sible, J.C. Dissection of the XChk1 Signaling Pathway in Xenopus Laevis Embryos. Mol. Biol. Cell 2000, 11, 3101–3108. [Google Scholar] [CrossRef]
- Conn, C.W.; Lewellyn, A.L.; Maller, J.L. The DNA Damage Checkpoint in Embryonic Cell Cycles Is Dependent on the DNA-to-Cytoplasmic Ratio. Dev. Cell 2004, 7, 275–281. [Google Scholar] [CrossRef]
- Ohkumo, T.; Masutani, C.; Eki, T.; Hanaoka, F. Deficiency of the Caenorhabditis Elegans DNA Polymerase Eta Homologue Increases Sensitivity to UV Radiation during Germ-Line Development. Cell Struct. Funct. 2006, 31, 29–37. [Google Scholar] [CrossRef][Green Version]
- Tada, S.; Li, A.; Maiorano, D.; Mechali, M.; Blow, J.J. Repression of Origin Assembly in Metaphase Depends on Inhibition of RLF- B/Cdt1 by Geminin. Nat. Cell Biol. 2001, 3, 107–113. [Google Scholar] [CrossRef]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Dutta, A. Inhibition of Eukaryotic DNA Replication by Geminin Binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Kaneko, K.J.; Pan, H.; DePamphilis, M.L. Geminin Is Essential to Prevent DNA Re-Replication-Dependent Apoptosis in Pluripotent Cells, but Not in Differentiated Cells. Stem Cells 2015, 33, 3239–3253. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Vinson, R.K.; Hales, B.F. DNA Repair during Organogenesis. Mutat. Res. 2002, 509, 79–91. [Google Scholar] [CrossRef]
- Ahmadi, A.; Ng, S.C. Developmental Capacity of Damaged Spermatozoa. Hum. Reprod. 1999, 14, 2279–2285. [Google Scholar] [CrossRef] [PubMed]
- Evenson, D.; Jost, L. Sperm Chromatin Structure Assay Is Useful for Fertility Assessment. Methods Cell Sci. Off. J. Soc. In Vitro Biol. 2000, 22, 169–189. [Google Scholar] [CrossRef]
- Fernández-Díez, C.; González-Rojo, S.; Lombó, M.; Herráez, M.P. Tolerance to Paternal Genotoxic Damage Promotes Survival during Embryo Development in Zebrafish (Danio Rerio). Biol. Open 2018, 7. [Google Scholar] [CrossRef]
- Blasco, M.A.; Serrano, M.; Fernandez-Capetillo, O. Genomic Instability in IPS: Time for a Break. EMBO J. 2011, 30, 991–993. [Google Scholar] [CrossRef]
- Gurtu, V.E.; Verma, S.; Grossmann, A.H.; Liskay, R.M.; Skarnes, W.C.; Baker, S.M. Maternal Effect for DNA Mismatch Repair in the Mouse. Genetics 2002, 160, 271–277. [Google Scholar]
- Fan, X.; Li, Y.; Zhang, Y.; Sang, M.; Cai, J.; Li, Q.; Ozaki, T.; Ono, T.; He, D. High Mutation Levels Are Compatible with Normal Embryonic Development in Mlh1-Deficient Mice. Radiat. Res. 2016, 186, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Schramm, R.D.; Latham, K.E. Developmental Regulation and in Vitro Culture Effects on Expression of DNA Repair and Cell Cycle Checkpoint Control Genes in Rhesus Monkey Oocytes and Embryos. Biol. Reprod. 2005, 72, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Kasparek, T.R.; Humphrey, T.C. DNA Double-Strand Break Repair Pathways, Chromosomal Rearrangements and Cancer. Semin. Cell Dev. Biol. 2011, 22, 886–897. [Google Scholar] [CrossRef]
- Lim, D.S.; Hasty, P. A Mutation in Mouse Rad51 Results in an Early Embryonic Lethal That Is Suppressed by a Mutation in P53. Mol. Cell. Biol. 1996, 16, 7133–7143. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.-L.; Shen, X.-H.; Shuang, L.; Kwon, J.-W.; Seong, M.-J.; Kim, N.-H. Inhibition of DNA Repair Protein RAD51 Affects Porcine Preimplantation Embryo Development. Reproduction 2019, 157, 223–234. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 Protects Nascent DNA from Mre11-Dependent Degradation and Promotes Continuous DNA Synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef]
- O’Farrell, P.H.; Stumpff, J.; Su, T.T. Embryonic Cleavage Cycles: How Is a Mouse like a Fly? Curr. Biol. 2004, 14, R35–R45. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome Instability Is Common in Human Cleavage-Stage Embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Munné, S.; Alikani, M.; Tomkin, G.; Grifo, J.; Cohen, J. Embryo Morphology, Developmental Rates, and Maternal Age Are Correlated with Chromosome Abnormalities. Fertil. Steril. 1995, 64, 382–391. [Google Scholar]
- Christodoulou, C.; Dheedene, A.; Heindryckx, B.; van Nieuwerburgh, F.; Deforce, D.; De Sutter, P.; Menten, B.; Van den Abbeel, E. Preimplantation Genetic Diagnosis for Chromosomal Rearrangements with the Use of Array Comparative Genomic Hybridization at the Blastocyst Stage. Fertil. Steril. 2017, 107, 212–219.e3. [Google Scholar] [CrossRef][Green Version]
- Reijns, M.A.M.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic Removal of Ribonucleotides from DNA Is Essential for Mammalian Genome Integrity and Development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef]
- Uehara, R.; Cerritelli, S.M.; Hasin, N.; Sakhuja, K.; London, M.; Iranzo, J.; Chon, H.; Grinberg, A.; Crouch, R.J. Two RNase H2 Mutants with Differential RNMP Processing Activity Reveal a Threshold of Ribonucleotide Tolerance for Embryonic Development. Cell Rep. 2018, 25, 1135–1145.e5. [Google Scholar] [CrossRef]
- McNairn, A.J.; Chuang, C.-H.; Bloom, J.C.; Wallace, M.D.; Schimenti, J.C. Female-Biased Embryonic Death from Inflammation Induced by Genomic Instability. Nature 2019, 567, 105–108. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Vander Griend, D.J.; Antony, L.; Dalrymple, S.; De Marzo, A.M.; Drake, C.G.; Isaacs, J.T. Androgen Receptor as a Licensing Factor for DNA Replication in Androgen-Sensitive Prostate Cancer Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15085–15090. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 Acts at Stalled Replication Forks to Prevent Interferon Induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Pera, M.F. Stem Cells: The Dark Side of Induced Pluripotency. Nature 2011, 471, 46–47. [Google Scholar] [CrossRef]
- Chun, C.; Wu, Y.; Lee, S.-H.; Williamson, E.A.; Reinert, B.L.; Jaiswal, A.S.; Nickoloff, J.A.; Hromas, R.A. The Homologous Recombination Component EEPD1 Is Required for Genome Stability in Response to Developmental Stress of Vertebrate Embryogenesis. Cell Cycle 2016, 15, 957–962. [Google Scholar] [CrossRef]
- Juan, H.C.; Lin, Y.; Chen, H.R.; Fann, M.J. Cdk12 Is Essential for Embryonic Development and the Maintenance of Genomic Stability. Cell Death Differ. 2016, 23, 1038–1048. [Google Scholar] [CrossRef]
- Delhanty, J.D.; Harper, J.C.; Ao, A.; Handyside, A.H.; Winston, R.M. Multicolour FISH Detects Frequent Chromosomal Mosaicism and Chaotic Division in Normal Preimplantation Embryos from Fertile Patients. Hum. Genet. 1997, 99, 755–760. [Google Scholar] [CrossRef]
- Bolton, H.; Graham, S.J.L.; Van der Aa, N.; Kumar, P.; Theunis, K.; Fernandez Gallardo, E.; Voet, T.; Zernicka-Goetz, M. Mouse Model of Chromosome Mosaicism Reveals Lineage-Specific Depletion of Aneuploid Cells and Normal Developmental Potential. Nat. Commun. 2016, 7, 11165. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.J.; Verwiel, E.T.P.; Heijdra, Y.; Vulliamy, T.; Kamping, E.J.; Hehir-Kwa, J.Y.; Bongers, E.M.H.F.; Pfundt, R.; van Emst, L.; van Leeuwen, F.N.; et al. Revertant Somatic Mosaicism by Mitotic Recombination in Dyskeratosis Congenita. Am. J. Hum. Genet. 2012, 90, 426–433. [Google Scholar] [CrossRef]
- Lesly, S.; Bandura, J.L.; Calvi, B.R. Rapid DNA Synthesis During Early Drosophila Embryogenesis Is Sensitive to Maternal Humpty Dumpty Protein Function. Genetics 2017, 207, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.J.; Bicknell, L.S.; Carroll, P.; Higgs, M.R.; Shaheen, R.; Murray, J.E.; Papadopoulos, D.K.; Leitch, A.; Murina, O.; Tarnauskaitė, Ž.; et al. Mutations in DONSON Disrupt Replication Fork Stability and Cause Microcephalic Dwarfism. Nat. Genet. 2017, 49, 537–549. [Google Scholar] [CrossRef]
- Evrony, G.D.; Cordero, D.R.; Shen, J.; Partlow, J.N.; Yu, T.W.; Rodin, R.E.; Hill, R.S.; Coulter, M.E.; Lam, A.-T.N.; Jayaraman, D.; et al. Integrated Genome and Transcriptome Sequencing Identifies a Noncoding Mutation in the Genome Replication Factor DONSON as the Cause of Microcephaly-Micromelia Syndrome. Genome Res. 2017, 27, 1323–1335. [Google Scholar] [CrossRef]
- Liu, P.; Yuan, B.; Carvalho, C.M.B.; Wuster, A.; Walter, K.; Zhang, L.; Gambin, T.; Chong, Z.; Campbell, I.M.; Coban Akdemir, Z.; et al. An Organismal CNV Mutator Phenotype Restricted to Early Human Development. Cell 2017, 168, 830–842.e7. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA Damage Response as a Candidate Anti-Cancer Barrier in Early Human Tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An Embryonic Stem Cell-like Gene Expression Signature in Poorly Differentiated Aggressive Human Tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Loeb, L.A. Mutator Phenotype May Be Required for Multistage Carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Checkpoint | Fast Cleaving Embryos | Mouse | Primates | Human |
|---|---|---|---|---|
| G1/S | − | − | n.d. | + |
| S | − | + | n.d. | + |
| G2/M | + | + | + | + |
| SAC | +/− | + | n.d. | + |
| DNA damage checkpoint activation | 32-cells (Xenopus laevis, [13]) | 2-cells | 4–8 cells | 4–8 cells |
| ZGA | 3000–6000 cells | 2-cells | 4–8 cells | 4–8 cells |
| Features | Embryos | Normal Cells | Cancer Cells |
|---|---|---|---|
| High proliferation rate | YES | NO | YES |
| Contact inhibition | NO | YES | NO |
| Migration | YES | NO | YES |
| Undifferentiated state | YES | NO | YES |
| Unstructured chromatin | YES | NO | YES/NO |
| Genetic instability (CNV) | YES | NO | YES |
| Inhibition of apoptosis | YES | NO | YES |
| Resistance to DNA damage | YES | NO | YES |
| Mutator phenotype | ? | NO | YES |
| N/C ratio | Low | High | Low/High |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kermi, C.; Aze, A.; Maiorano, D. Preserving Genome Integrity during the Early Embryonic DNA Replication Cycles. Genes 2019, 10, 398. https://doi.org/10.3390/genes10050398
Kermi C, Aze A, Maiorano D. Preserving Genome Integrity during the Early Embryonic DNA Replication Cycles. Genes. 2019; 10(5):398. https://doi.org/10.3390/genes10050398
Chicago/Turabian StyleKermi, Chames, Antoine Aze, and Domenico Maiorano. 2019. "Preserving Genome Integrity during the Early Embryonic DNA Replication Cycles" Genes 10, no. 5: 398. https://doi.org/10.3390/genes10050398
APA StyleKermi, C., Aze, A., & Maiorano, D. (2019). Preserving Genome Integrity during the Early Embryonic DNA Replication Cycles. Genes, 10(5), 398. https://doi.org/10.3390/genes10050398