Comparison of Bacterial Populations in the Ceca of Swine at Two Different Stages and Their Functional Annotations

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Ethics Statement, Animal Rearing, and Feeding
2.2. Sample Collection, Nucleic Acid Extraction, and 16s rRNA High-Throughput Sequencing
2.3. Sequence Quality, Assembly, Preprocessing, and Clustering
2.4. Taxonomic Assignment and Diversity Analysis
2.5. Bioinformatics Analysis for Functional Annotations
2.5.1. Multivariate Statistical Analysis to Explore Complex Ecological Associations
2.5.2. Comparative Functional Annotation of Cecum Content
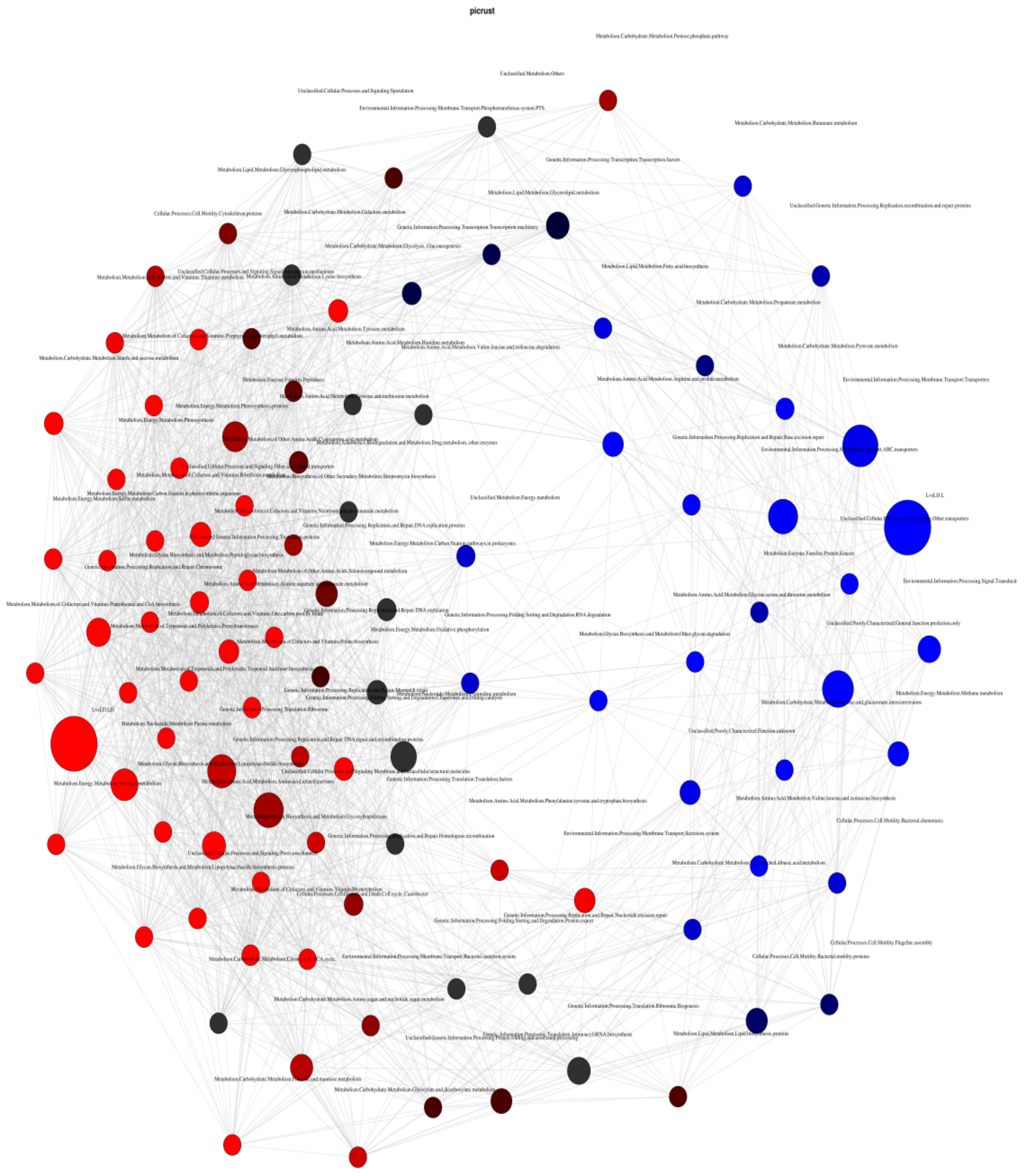
2.5.3. Network Analysis through Pearson’s Correlation
3. Results
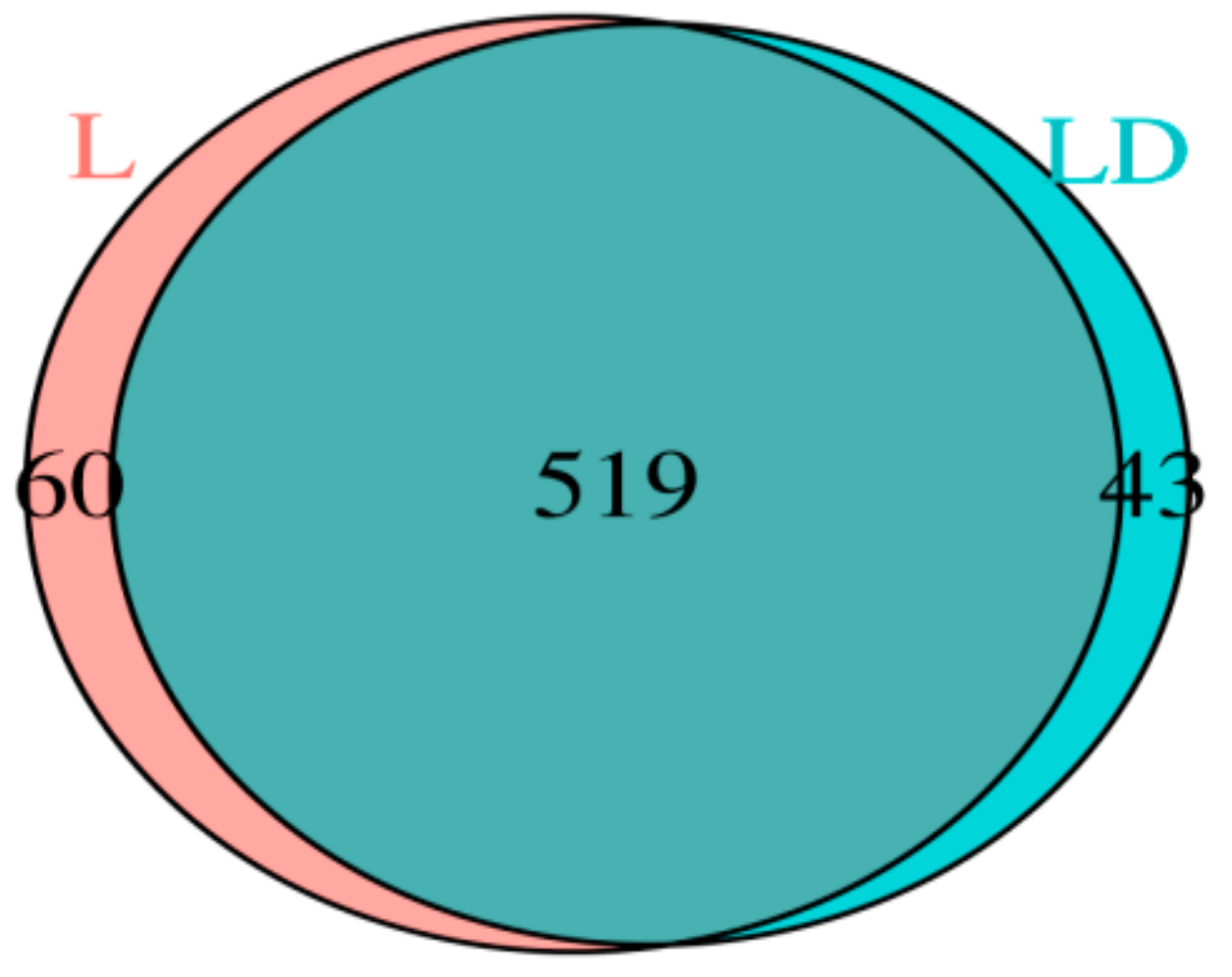
3.1. Sequence Analysis of Cecum Content
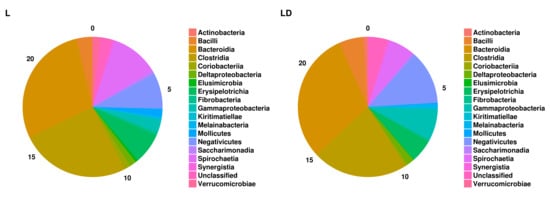
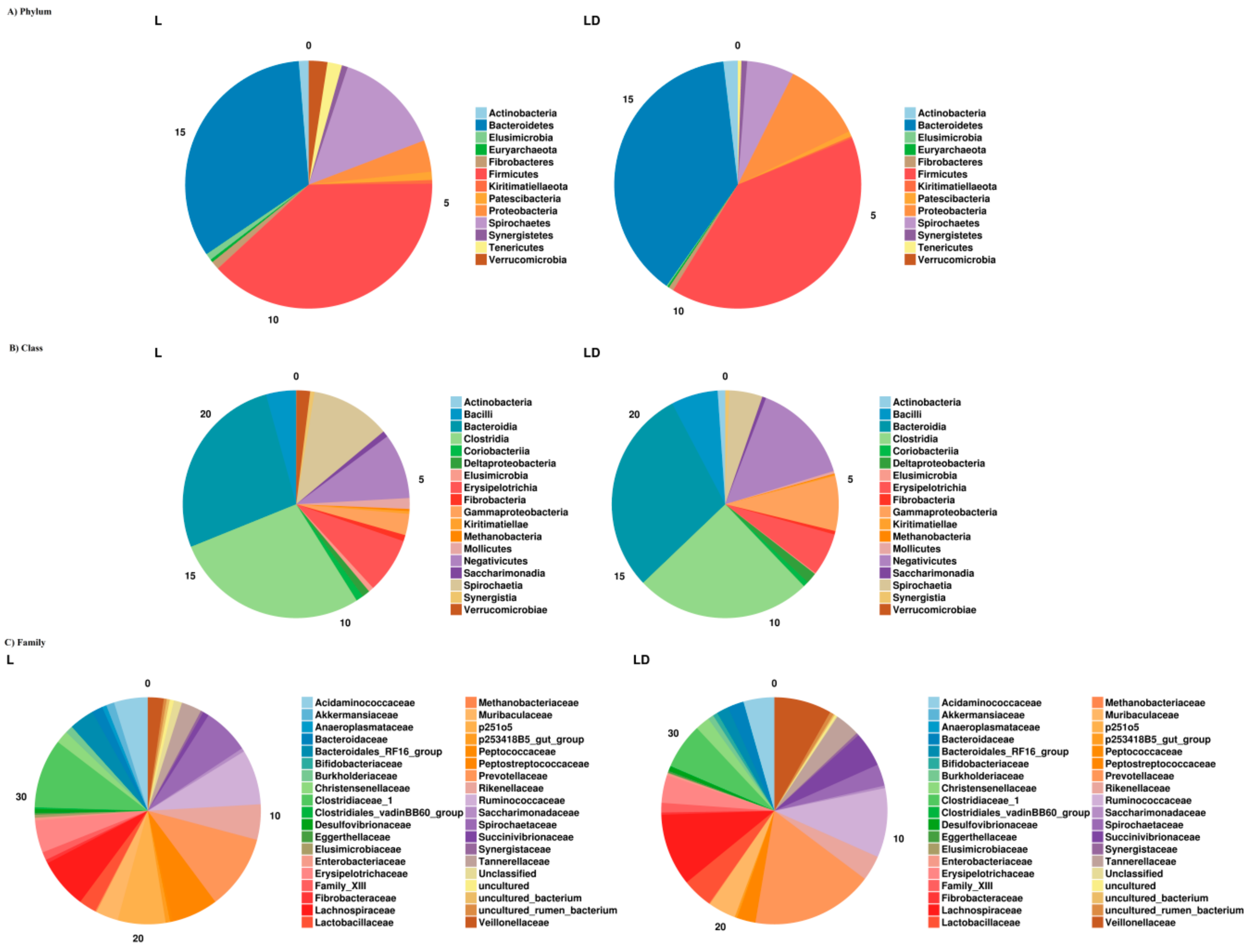
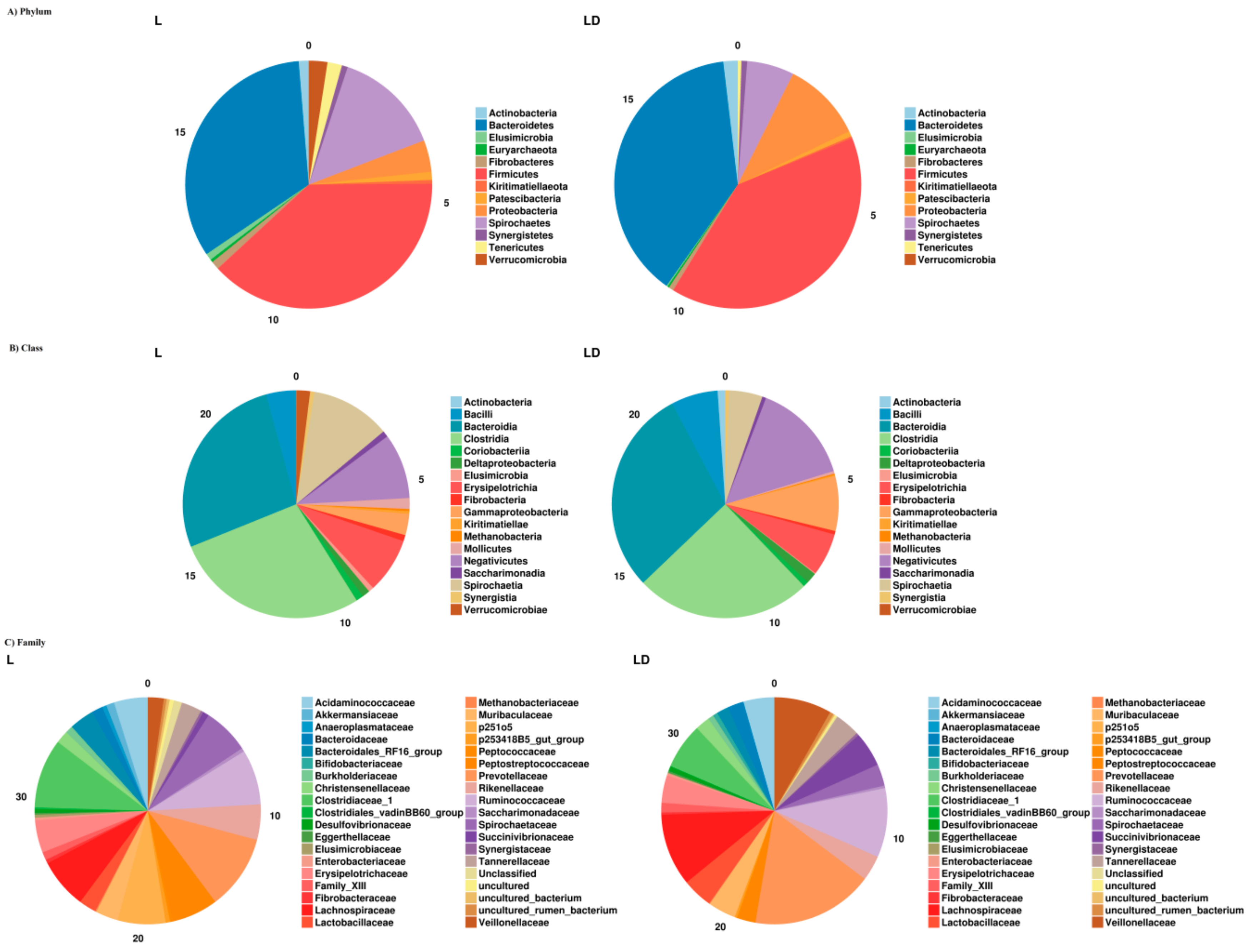
3.2. Taxonomic Assignment
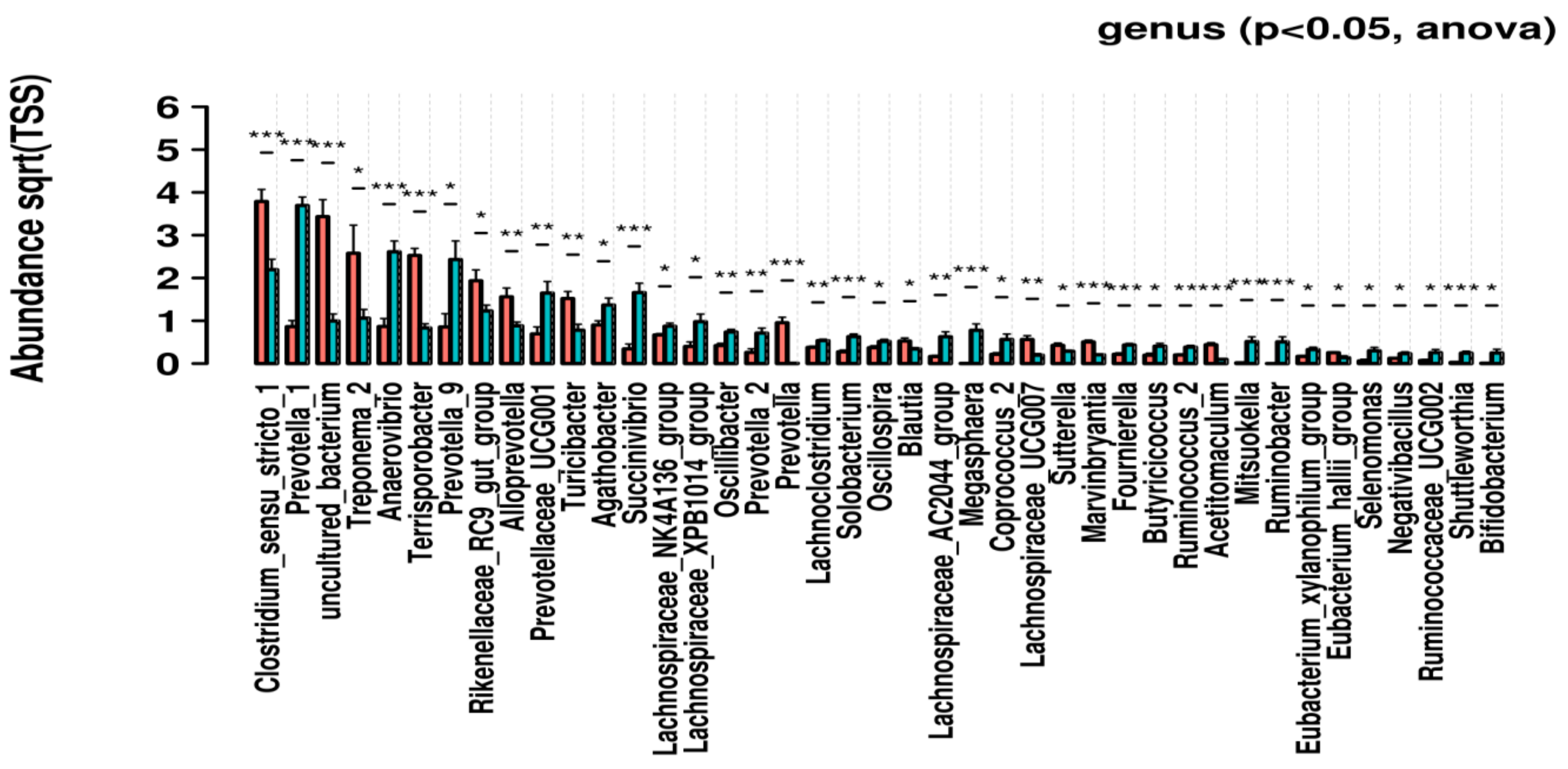
3.3. Genus-Level Univariate Analysis
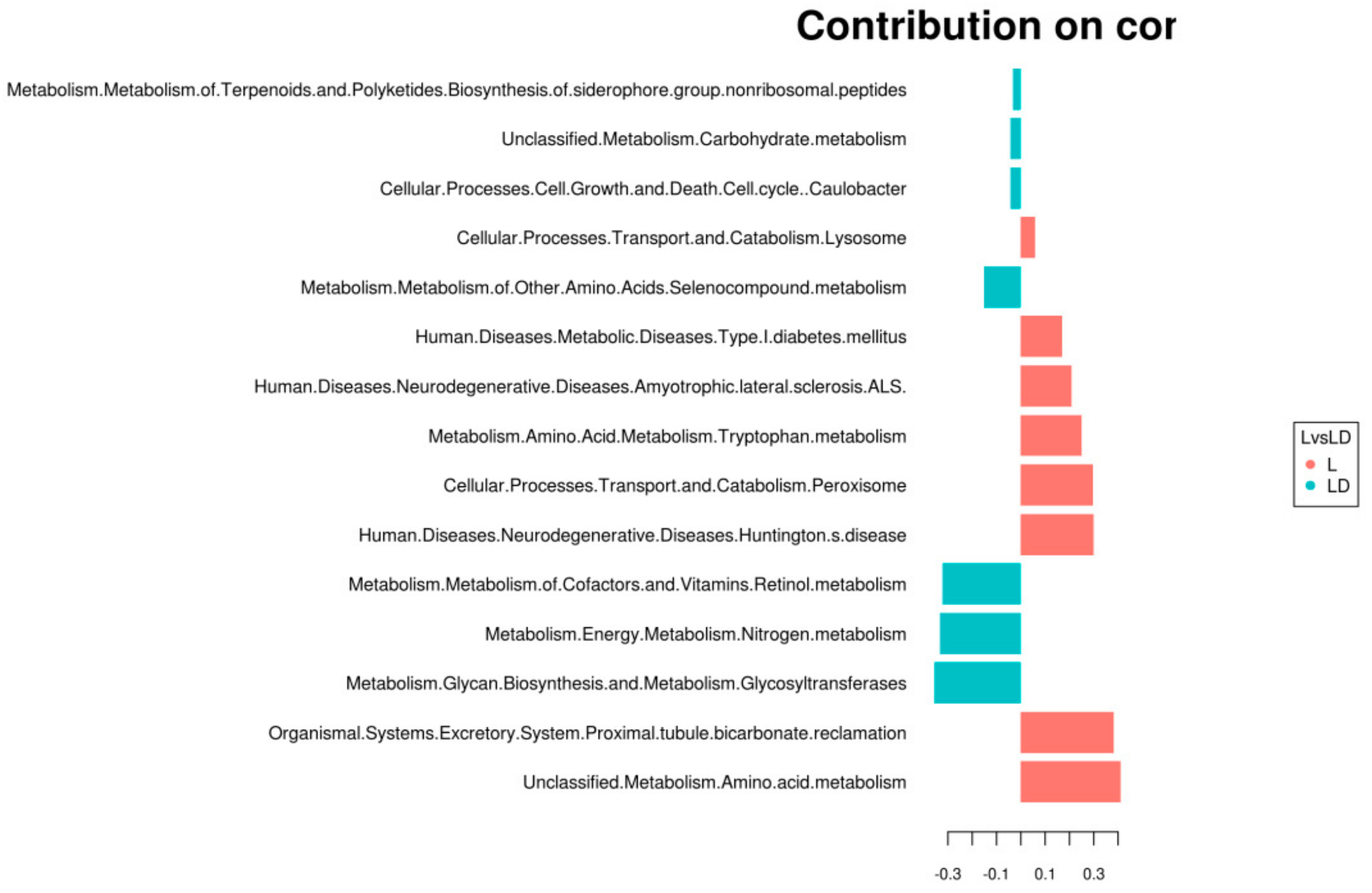
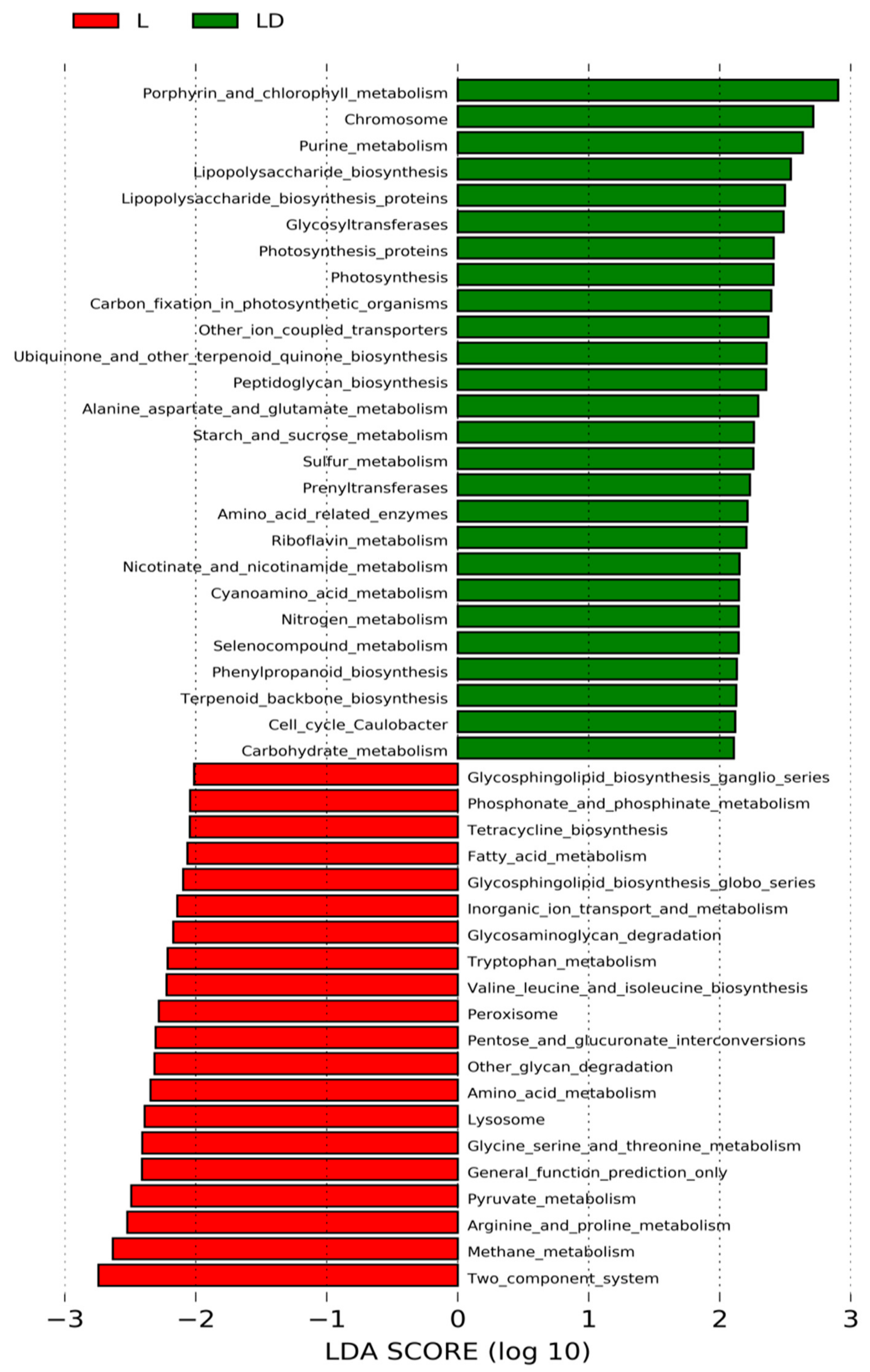
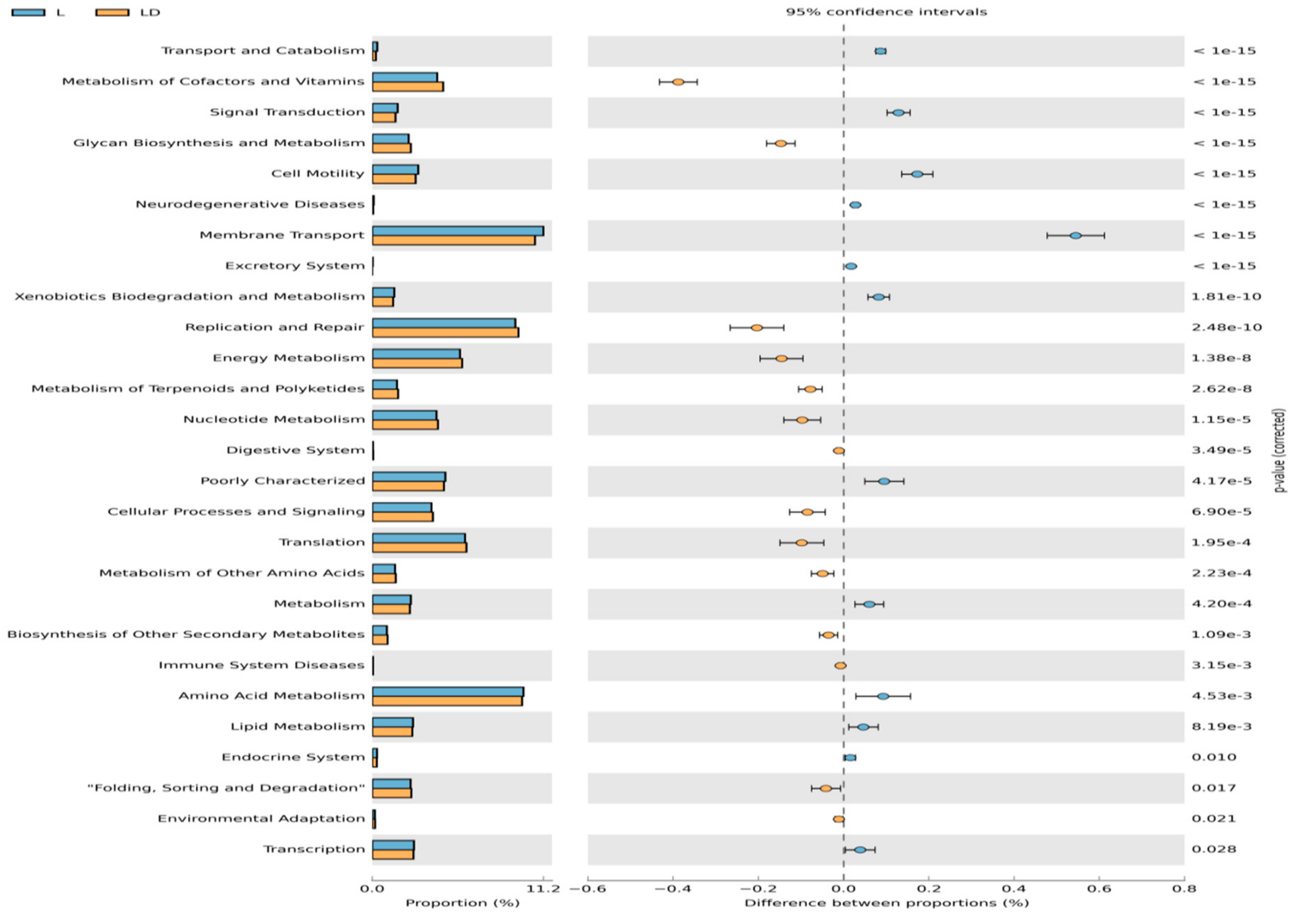
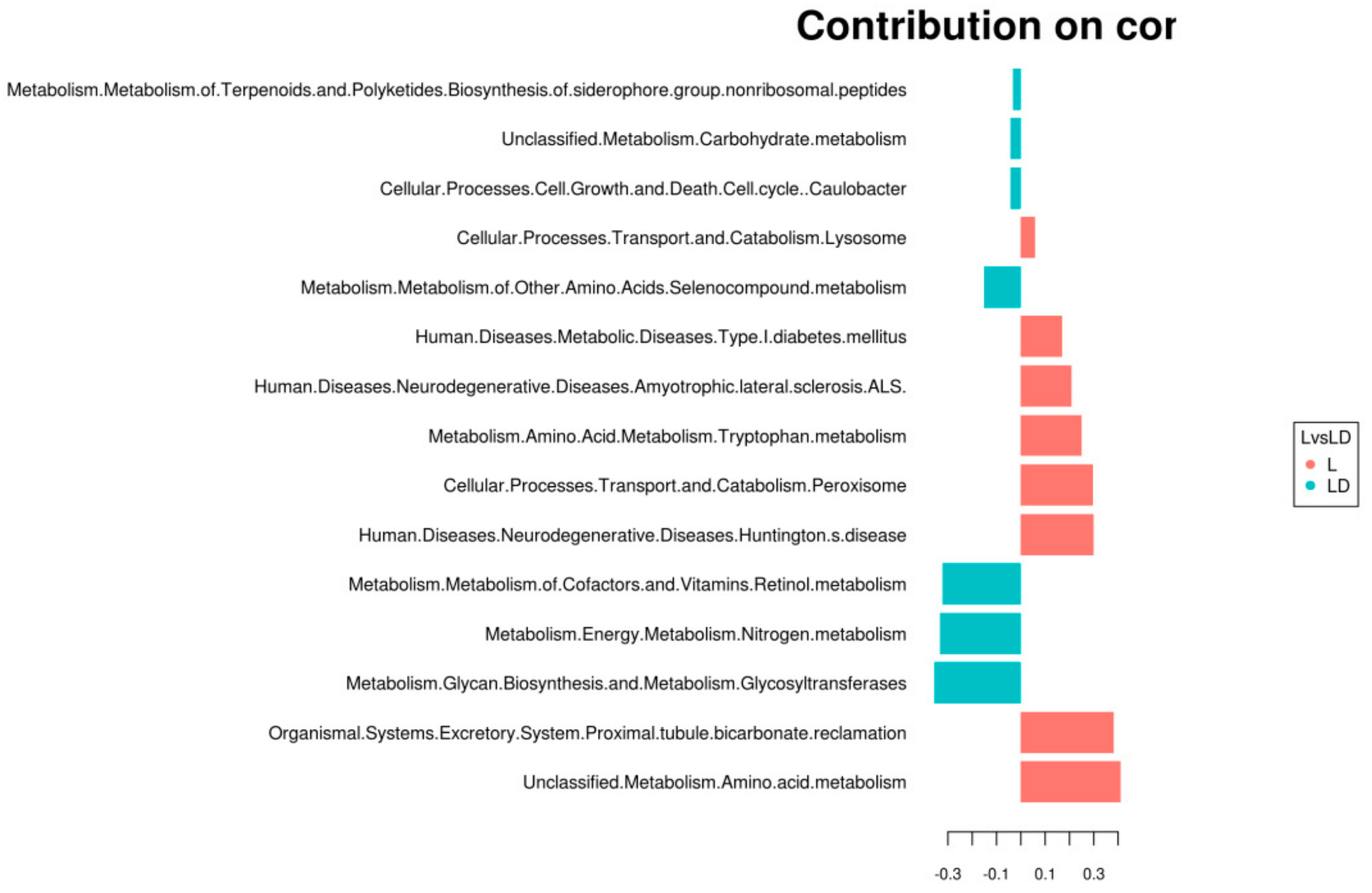
3.4. Statistical Analysis for Functional Annotation
4. Discussion
5. Conclusions
Nucleotide Sequence Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lamendella, R.; Santo Domingo, J.W.; Ghosh, S.; Martinson, J.; Oerther, D.B. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 103. [Google Scholar] [CrossRef]
- Isaacson, R.; Kim, H.B. The intestinal microbiome of the pig. Anim. Health Res. Rev. 2012, 13, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Angenent, L.T.; Gordon, J.I. Getting a grip on things: How do communities of bacterial symbionts become established in our intestine? Nat. Immunol. 2004, 5, 569–573. [Google Scholar] [CrossRef]
- Looft, T.; Allen, H.K.; Cantarel, B.L.; Levine, U.Y.; Bayles, D.O.; Alt, D.P.; Henrissat, B.; Stanton, T.B. Bacteria, phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014, 8, 1566–1576. [Google Scholar] [CrossRef]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek 1998, 73, 127–141. [Google Scholar] [CrossRef]
- Strathdee, F.; Free, A. Denaturing Gradient Gel Electrophoresis (DGGE). In DNA Electrophoresis: Methods and Protocols; Makovets, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 145–157. [Google Scholar]
- Marsh, T.L. Terminal restriction fragment length polymorphism (T-RFLP): An emerging method for characterizing diversity among homologous populations of amplification products. Curr. Opin. Microbiol. 1999, 2, 323–327. [Google Scholar] [CrossRef]
- Cornejo, P.; Azcon-Aguilar, C.; Miguel Barea, J.; Ferrol, N. Temporal temperature gradient gel electrophoresis (TTGE) as a tool for the characterization of arbuscular mycorrhizal fungi. FEMS Microbiol. Lett. 2004, 241, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ronaghi, M.; Uhlén, M.; Nyrén, P. A sequencing method based on real-time pyrophosphate. Science 1998, 281, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Zoetendal, E.G.; Collier, C.T.; Koike, S.; Mackie, R.I.; Gaskins, H.R. Molecular ecological analysis of the gastrointestinal microbiota: A review. J. Nutr. 2004, 134, 465–472. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 2011, 153, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.S.; Baker, B.J.; Thomas, B.C.; Singer, S.W.; Banfield, J.F. EMIRGE: Reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011, 12, R44. [Google Scholar] [CrossRef]
- Yuan, C.; Lei, J.; Cole, J.; Sun, Y. Reconstructing 16S rRNA genes in metagenomic data. Bioinformatics 2015, 31, i35–i43. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Tang, H.; Doak, T.G.; Ye, Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. In Biocomputing 2011; World Scientific: Kohala Coast, HI, USA, 2011; pp. 165–176. [Google Scholar]
- Albanese, D.; Fontana, P.; De Filippo, C.; Cavalieri, D.; Donati, C. MICCA: A complete and accurate software for taxonomic profiling of metagenomic data. Sci. Rep. 2015, 5, 9743. [Google Scholar] [CrossRef]
- Fiannaca, A.; La Paglia, L.; La Rosa, M.; Renda, G.; Rizzo, R.; Gaglio, S.; Urso, A. Deep learning models for bacteria taxonomic classification of metagenomic data. BMC Bioinform. 2018, 19, 198. [Google Scholar] [CrossRef]
- Maltecca, C.; Lu, D.; Schillebeeckx, C.; McNulty, N.; Schawb, C.; Schull, C.; Tiezzi, F. Predicting Growth and Carcass Traits in Swine Using Metagenomic Data and Machine Learning Algorithms. bioRxiv 2018, 363309. [Google Scholar]
- Saraswati, S.; Sitaraman, R. Aging and the human gut microbiota—From correlation to causality. Front. Microbiol. 2015, 5, 764. [Google Scholar] [CrossRef]
- Hu, J.; Nie, Y.; Chen, J.; Zhang, Y.; Wang, Z.; Fan, Q.; Yan, X. Gradual changes of gut microbiota in weaned miniature piglets. Front. Microbiol. 2016, 7, 1727. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I. Principal component analysis. In International Encyclopedia of Statistical Science; Springer-Verlag: Heidelberg, Alemania, 2011; pp. 1094–1096. [Google Scholar]
- Liu, Q. Variation partitioning by partial redundancy analysis (RDA). Environmetrics 1997, 8, 75–85. [Google Scholar] [CrossRef]
- Ter Braak, C.J. Canonical correspondence analysis: A new eigenvector technique for multivariate direct gradient analysis. Ecology 1986, 67, 1167–1179. [Google Scholar] [CrossRef]
- Hill, M.O.; Gauch, H.G. Detrended correspondence analysis: An improved ordination technique. Vegetatio 1880, 42, 47–58. [Google Scholar] [CrossRef]
- Denman, S.E.; Martinez Fernandez, G.; Shinkai, T.; Mitsumori, M.; McSweeney, C.S. Metagenomic analysis of the rumen microbial community following inhibition of methane formation by a halogenated methane analog. Front. Microbiol. 2015, 6, 1087. [Google Scholar] [CrossRef]
- Langille, M.G.; Meehan, C.J.; Koenig, J.E.; Dhanani, A.S.; Rose, R.A.; Howlett, S.E.; Beiko, R.G. Microbial shifts in the aging mouse gut. Microbiome 2014, 2, 50. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C. SEARCH_16S: A new algorithm for identifying 16S ribosomal RNA genes in contigs and chromosomes. bioRxiv 2017, 124131. [Google Scholar]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Santo Domingo, J.; Shanks, O.C. Identification of chicken-specific fecal microbial sequences using a metagenomic approach. Water Res. 2007, 41, 3561–3574. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Hao, X.; Jiang, R.; Chen, T. Clustering 16S rRNA for OTU prediction: A method of unsupervised Bayesian clustering. Bioinformatics 2011, 27, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Plummer, E.; Twin, J.; Bulach, D.M.; Garland, S.M.; Tabrizi, S.N. A comparison of three bioinformatics pipelines for the analysis of preterm gut microbiota using 16S rRNA gene sequencing data. J. Proteom. Bioinform. 2015, 8, 283. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R. UCHIME2: Improved chimera prediction for amplicon sequencing. BioRxiv 2016, 074252. [Google Scholar]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. GigaScience 2012, 1, 7. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2009, 26, 266–267. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Holland, S.M. Non-Metric Multidimensional Scaling (MDS); Department of Geology, University of Georgia: Athens, Greece, 2008. [Google Scholar]
- Le Cao, K.-A.; Costello, M.-E.; Lakis, V.A.; Bartolo, F.; Chua, X.-Y.; Brazeilles, R.; Rondeau, P. MixMC: A multivariate statistical framework to gain insight into microbial communities. PLoS ONE 2016, 11, e0160169. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef]
- Cuevas, A.; Febrero, M.; Fraiman, R. An anova test for functional data. Comput. Stat. Data Anal. 2004, 47, 111–122. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Kreimer, A.; Meilijson, I.; Gophna, U.; Sharan, R.; Ruppin, E. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res. 2010, 38, 3857–3868. [Google Scholar] [CrossRef]
- Joyce, S.A.; Gahan, C.G. The gut microbiota and the metabolic health of the host. Curr. Opin. Gastroenterol. 2014, 30, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef]
- Allen, H.K.; Looft, T.; Bayles, D.O.; Humphrey, S.; Levine, U.Y.; Alt, D.; Stanton, T.B. Antibiotics in feed induce prophages in swine fecal microbiomes. MBio 2011, 2, e00260-11. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.J.; Robinson, I.; Bucklin, J.; Booth, G. Comparison of bacterial populations of the pig cecum and colon based upon enumeration with specific energy sources. Appl. Environ. Microbiol. 1979, 37, 1142–1151. [Google Scholar] [PubMed]
- Kim, H.B.; Isaacson, R.E. The pig gut microbial diversity: Understanding the pig gut microbial ecology through the next generation high throughput sequencing. Vet. Microbiol. 2015, 177, 242–251. [Google Scholar] [CrossRef]
- Pajarillo, E.A.B.; Chae, J.-P.; Balolong, M.P.; Kim, H.B.; Kang, D.-K. Assessment of fecal bacterial diversity among healthy piglets during the weaning transition. J. Gen. Appl. Microbiol. 2014, 60, 140–146. [Google Scholar] [CrossRef]
- Konstantinov, S.R.; Favier, C.F.; Zhu, W.Y.; Williams, B.A.; Klüß, J.; Souffrant, W.-B.; de Vos, W.M.; Akkermans, A.D.; Smidt, H. Microbial diversity studies of the porcine gastrointestinal ecosystem during weaning transition. Anim. Res. 2004, 53, 317–324. [Google Scholar] [CrossRef]
- Mackie, R.I.; Sghir, A.; Gaskins, H.R. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 1999, 69, 1035s–1045s. [Google Scholar] [CrossRef]
- Poroyko, V.; White, J.R.; Wang, M.; Donovan, S.; Alverdy, J.; Liu, D.C.; Morowitz, M.J. Gut microbial gene expression in mother-fed and formula-fed piglets. PLoS ONE 2010, 5, e12459. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Joice, R.; Yasuda, K.; Shafquat, A.; Morgan, X.C.; Huttenhower, C. Determining microbial products and identifying molecular targets in the human microbiome. Cell Metab. 2014, 20, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.J.; Kang, H.-S.; Chu, J.; Huang, Y.-H.; Gordon, E.A.; Reddy, B.V.B.; Ternei, M.A.; Craig, J.W.; Brady, S.F. Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc. Natl. Acad. Sci. USA 2015, 112, E4825–E4834. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Oakeson, K.F.; Orr, T.J.; Miller, A.W.; Forbey, J.S.; Phillips, C.D.; Dale, C.; Weiss, R.B.; Dearing, M.D. Metagenomic sequencing provides insights into the location of microbial detoxification in the gut of a small mammalian herbivore. bioRxiv 2018, 299198. [Google Scholar]
- Virkel, G.; Ballent, M.; Lanusse, C.; Lifschitz, A. Role of ABC transporters in veterinary medicine: Pharmaco-toxicological implications. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Yang, H.; Huang, X.; Fang, S.; He, M.; Zhao, Y.; Wu, Z.; Yang, M.; Zhang, Z.; Chen, C.; Huang, L. Unraveling the fecal microbiota and metagenomic functional capacity associated with feed efficiency in pigs. Front. Microbiol. 2017, 8, 1555. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lingner, T.; Meinicke, P.; Tech, M. Orphelia: Predicting genes in metagenomic sequencing reads. Nucleic Acids Res. 2009, 37, W101–W105. [Google Scholar] [CrossRef]
- Hoff, K.J.; Tech, M.; Lingner, T.; Daniel, R.; Morgenstern, B.; Meinicke, P. Gene prediction in metagenomic fragments: A large scale machine learning approach. BMC Bioinform. 2008, 9, 217. [Google Scholar] [CrossRef]
- El Allali, A.; Rose, J.R. MGC: A metagenomic gene caller. BMC Bioinform. 2013, 14, S6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, J.; Hu, G.; Zhu, H. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinform. 2013, 14, S12. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Huang, X.; Fang, S.; Xin, W.; Huang, L.; Chen, C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci. Rep. 2016, 6, 27427. [Google Scholar] [CrossRef]
- Kouchiwa, T.; Wada, K.; Uchiyama, M.; Kasezawa, N.; Niisato, M.; Murakami, H.; Fukuyama, K.; Yokogoshi, H. Age-related changes in serum amino acids concentrations in healthy individuals. Clin. Chem. Lab. Med. 2012, 50, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Yang, T.; Wang, Y.; Xing, K.; Zhang, F.; Zhao, X.; Ao, H.; Chen, S.; Liu, J.; Wang, C. Metagenomic analysis of cecal microbiome identified microbiota and functional capacities associated with feed efficiency in landrace finishing pigs. Front. Microbiol. 2017, 8, 1546. [Google Scholar] [CrossRef]
- Mach, N.; Berri, M.; Estellé, J.; Levenez, F.; Lemonnier, G.; Denis, C.; Leplat, J.J.; Chevaleyre, C.; Billon, Y.; Doré, J. Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ. Microbiol. Rep. 2015, 7, 554–569. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Mach, N.; Lepage, P.; Levenez, F.; Denis, C.; Lemonnier, G.; Leplat, J.-J.; Billon, Y.; Berri, M.; Doré, J.; et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J. 2016, 10, 2973–2977. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Protein | Crude Fat | Ca | P | Crude Fiber | Crude Ash | Lysine | DE | DCP |
|---|---|---|---|---|---|---|---|---|---|
| Stage 1 (L) | 19.0 | 6.7 | 0.4 | 1.5 | 4.0 | 8.0 | 1.40 | 3.6 | 16.0 |
| Stage 2 (LD) | 14.5 | 5.5 | 0.4 | 1.2 | 7.0 | 8.0 | 0.80 | 3.5 | 12.0 |
| Sample Name | Sex | Total Bases | Read | GC (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| K4.4L | F | 86,303,564 | 189,866 | 53.08 | 98.54 | 95.24 |
| K4.10L | M | 100,659,045 | 223,531 | 53.1 | 98.56 | 95.22 |
| Y5.2L | F | 78,466,731 | 172,363 | 52.8 | 98.51 | 95.14 |
| Y5.3L | M | 79,260,896 | 175,057 | 53.03 | 98.55 | 95.26 |
| Y5.4L | F | 74,925,854 | 165,186 | 53.18 | 98.58 | 95.33 |
| Y5.6L | M | 85,490,768 | 189,176 | 53.11 | 98.52 | 95.06 |
| Y5.9L | M | 93,195,851 | 206,722 | 53.3 | 98.55 | 95.25 |
| Y5.12L | F | 92,420,536 | 204,127 | 53.21 | 98.63 | 95.43 |
| K4.5L.D | M | 70,633,514 | 155,507 | 53.32 | 98.62 | 95.35 |
| K4.8L.D | F | 77,651,573 | 170,983 | 53.21 | 98.62 | 95.47 |
| Y5.1L.D | F | 74,716,039 | 164,985 | 53.61 | 98.78 | 95.75 |
| Y5.5L.D | F | 81,054,520 | 178,286 | 53.31 | 98.78 | 95.8 |
| Y5.7L.D | F | 76,811,356 | 168,951 | 53.58 | 98.56 | 95.29 |
| Y5.8L.D | M | 85,996,517 | 188,902 | 53.44 | 98.54 | 95.29 |
| Y5.10L.D | M | 70,647,269 | 155,842 | 53.62 | 98.52 | 95.27 |
| Y5.11L.D | M | 62,782,136 | 138,382 | 53.68 | 98.65 | 95.46 |
| Sample Name | OTUs | Chao1 | Shannon | Simpson | Goods Coverage |
|---|---|---|---|---|---|
| K4.5LD | 349 | 399.833 | 5.999 | 0.961 | 0.995 |
| K4.8LD | 279 | 303.117 | 4.707 | 0.891 | 0.997 |
| K4.4L | 454 | 509.122 | 6.214 | 0.966 | 0.997 |
| K4.10L | 476 | 509.552 | 6.298 | 0.973 | 0.998 |
| Y5.1LD | 360 | 405.217 | 5.941 | 0.967 | 0.996 |
| Y5.5LD | 381 | 422.576 | 6.108 | 0.973 | 0.999 |
| Y5.7LD | 362 | 399.631 | 5.827 | 0.960 | 0.996 |
| Y5.8LD | 326 | 355.684 | 5.355 | 0.935 | 0.998 |
| Y5.10LD | 373 | 419.941 | 6.284 | 0.975 | 0.996 |
| Y5.11LD | 370 | 406.875 | 6.031 | 0.965 | 0.995 |
| Y5.2L | 418 | 456.478 | 5.756 | 0.958 | 0.997 |
| Y5.3L | 367 | 410.807 | 4.938 | 0.907 | 0.997 |
| Y5.4L | 382 | 429.275 | 6.051 | 0.973 | 0.997 |
| Y5.6L | 397 | 458.386 | 5.664 | 0.954 | 0.996 |
| Y5.9L | 310 | 351.437 | 4.998 | 0.923 | 0.998 |
| Y5.12L | 453 | 498.122 | 6.045 | 0.963 | 0.997 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, H.; Park, W.; Srikanth, K.; Choi, B.-H.; Cho, E.-S.; Lee, K.-T.; Kim, J.-M.; Kim, K.; Park, J.; Lim, D.; et al. Comparison of Bacterial Populations in the Ceca of Swine at Two Different Stages and Their Functional Annotations. Genes 2019, 10, 382. https://doi.org/10.3390/genes10050382
Kumar H, Park W, Srikanth K, Choi B-H, Cho E-S, Lee K-T, Kim J-M, Kim K, Park J, Lim D, et al. Comparison of Bacterial Populations in the Ceca of Swine at Two Different Stages and Their Functional Annotations. Genes. 2019; 10(5):382. https://doi.org/10.3390/genes10050382
Chicago/Turabian StyleKumar, Himansu, Woncheol Park, Krishnamoorthy Srikanth, Bong-Hwan Choi, Eun-Seok Cho, Kyung-Tai Lee, Jun-Mo Kim, Kwangmin Kim, Junhyung Park, Dajeong Lim, and et al. 2019. "Comparison of Bacterial Populations in the Ceca of Swine at Two Different Stages and Their Functional Annotations" Genes 10, no. 5: 382. https://doi.org/10.3390/genes10050382
APA StyleKumar, H., Park, W., Srikanth, K., Choi, B.-H., Cho, E.-S., Lee, K.-T., Kim, J.-M., Kim, K., Park, J., Lim, D., & Park, J.-E. (2019). Comparison of Bacterial Populations in the Ceca of Swine at Two Different Stages and Their Functional Annotations. Genes, 10(5), 382. https://doi.org/10.3390/genes10050382