AON-Mediated Exon Skipping to Bypass Protein Truncation in Retinal Dystrophies Due to the Recurrent CEP290 c.4723A > T Mutation. Fact or Fiction?

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Analysis
2.2. In Silico Analysis of the Nonsense c.4723A > T (p.Lys1575*) Mutation on Splicing
2.3. Cell Culture
2.4. AON and Transfection
2.5. RNA Analysis
2.6. Protein Analysis
2.7. Statistics
3. Results
3.1. Panel-Based Molecular Diagnosis Testing Identifies Homozygosity for the CEP290 c.4723A > T Founder Mutation in Two Individuals with Congenital Retinal Dystrophy of Variable Severity
3.2. In Silico Analysis Suggests That the CEP290 c.4723A > T Mutation Induced Nonsense-Associated Altered Splicing
3.3. mRNA Analysis Supports c.4723A > T-Mediated Nonsense-Associated Altered Splicing and Basal Endogenous Alternative Splicing of Exon 36
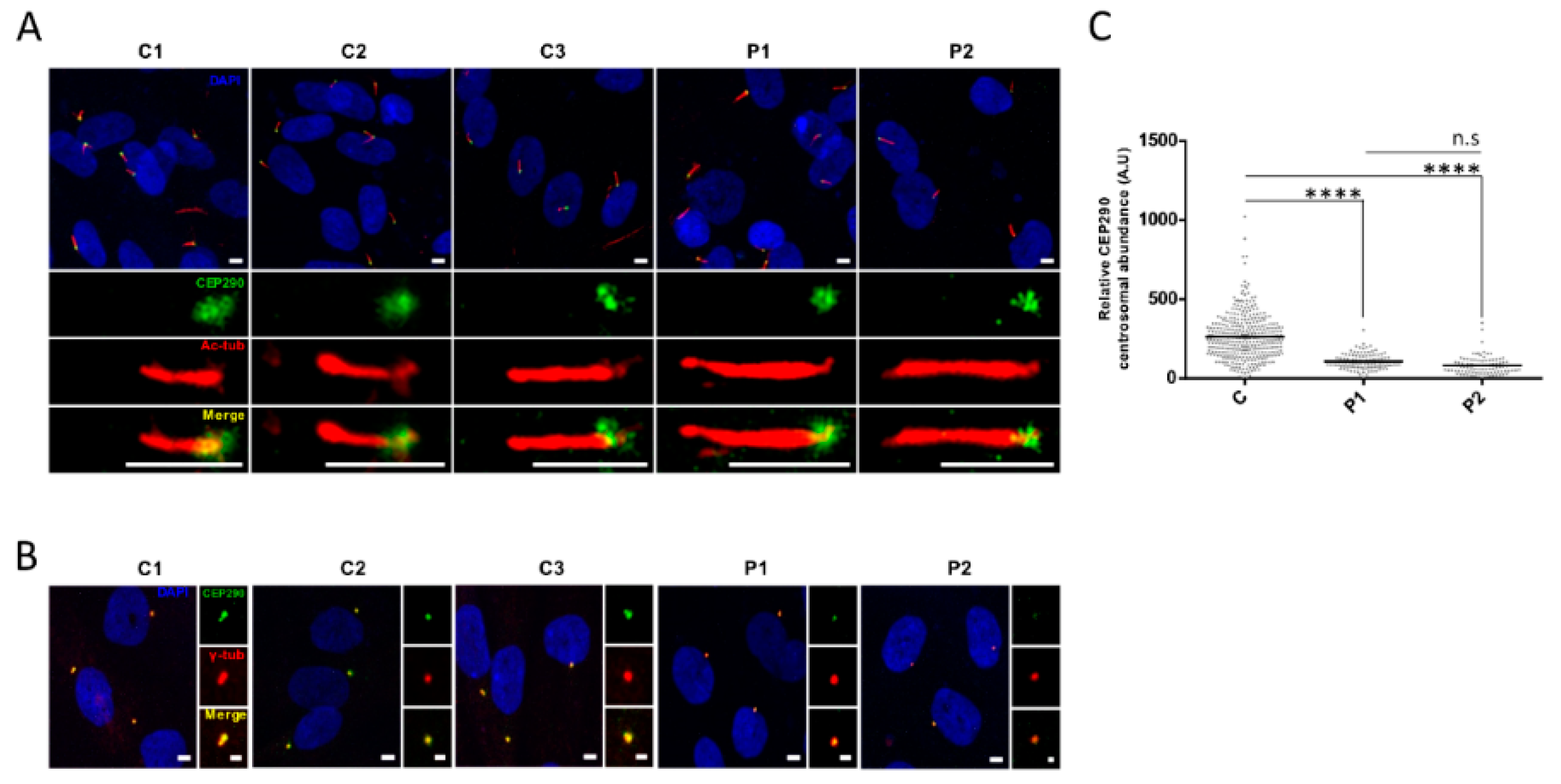
3.4. Protein Analysis Detects Low Levels of a CEP290 Protein That Localizes at the Centrosome in Patient Fibroblasts Homozygous for the c.4723A > T Nonsense Mutation
3.5. Cilia Analysis of Serum-Starved Patient Fibroblasts Shows Apparently Normal RAB8A Localization at the Centrosome but Elongated Axonemes
3.6. Targeting the Consensus Donor Splice Site Enables Dose- and Time-Dependent Skipping of CEP290 Exon 36
3.7. AON-Mediated Skipping Allows to Bypass Protein-Truncation but May Not Restore Full CEP290 Functions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaplan, J. Leber congenital amaurosis: From darkness to spotlight. Ophthalmic Genet. 2008, 29, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Hanein, S.; Perrault, I.; Gerber, S.; Tanguy, G.; Barbet, F.; Ducroq, D.; Calvas, P.; Dollfus, H.; Hamel, C.; Lopponen, T.; et al. Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype–phenotype correlations as a strategy for molecular diagnosis. Hum. Mutat. 2004, 23, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Craige, B.; Tsao, C.C.; Diener, D.R.; Hou, Y.; Lechtreck, K.F.; Rosenbaum, J.L.; Witman, G.B. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 2010, 190, 927–940. [Google Scholar] [CrossRef]
- Den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P.M. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Perrault, I.; Delphin, N.; Hanein, S.; Gerber, S.; Dufier, J.L.; Roche, O.; Defoort-Dhellemmes, S.; Dollfus, H.; Fazzi, E.; Munnich, A.; et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum. Mutat. 2007, 28, 416. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Aleman, T.S.; Jacobson, S.G.; Khanna, H.; Sumaroka, A.; Aquirre, G.K.; Schwartz, S.B.; Windsor, E.A.; He, S.; Chang, B.; et al. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: Implications for therapy of Leber congenital amaurosis. Hum. Mutat. 2007, 28, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Preising, M.N.; Schneider, U.; Friedburg, C.; Gruber, H.; Lindner, S. Lorenz, B. The Phenotypic spectrum of ophthalmic changes in CEP290 mutations. Klin. Monbl. Augenheilkd 2019, 236, 244–252. [Google Scholar]
- Gerard, G.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. AON-mediated exon skipping restores ciliation in fibroblasts harboring the common leber congenital amaurosis CEP290 mutation. Mol. Ther. Nucl. Acids 2012, 1, e29. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.; den Hollander, A.I.; van der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense oligonucleotide (AON)-based therapy for leber congenital amaurosis caused by a frequent mutation in CEP290. Mol. Ther. Nucl. Acids 2012, 1, e14. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.J.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell. Stem. 2016, 18, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Dulla, K.; Aguila, M.; Lane, A.; Jovanovic, K.; Parfitt, D.A.; Schulkens, I.; Chan, H.L.; Schmidt, I.; Beumer, W.; Vorthoren, L.; et al. Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c.2991+1655A>G LCA10 models. Mol. Ther. Nucl. Acids 2018, 12, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; Chung, D.C.; Duijkers, L.; Corral-Serrano, J.C.; Messchaert, M.; Xiao, R.; Bennett, J.; Vandenberghe, L.H.; Collin, R.W. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet. 2016, 25, 2552–2563. [Google Scholar]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Cideciyan, A.V.; Schwartz, S.B.; Wang, L.; Conlon, T.J.; Boye, S.L.; Flotte, T.R.; Byrne, B.J.; et al. Treatment of leber congenital amaurosis due to rpe65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther. 2008, 19, 979–990. [Google Scholar] [CrossRef]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and Efficacy of gene transfer for leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Mullins, R.F.; Dumitrescu, A.V.; Bhattarai, S.; Gratie, D.; Wang, K.; Stone, E.M.; Sheffield, V.; Drack, A.V. Subretinal gene therapy of mice with bardet-biedl syndrome type 1. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6118–6132. [Google Scholar] [CrossRef] [PubMed]
- Kloeckener-Gruissem, B.; Neidhart, J.; Magyar, I.; Plauchu, H.; Zech, J.C.; Morlé, L.; Palmer-Smith, S.M.; Macdonald, M.J.; Nas, V.; Fry, A.E.; et al. Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of Wagner syndrome. Eur. J. Hum. Genet. 2013, 21, 352–356. [Google Scholar] [CrossRef]
- Papon, J.F.; Perrault, I.; Coste, A.; Louis, B.; Gérard, X.; Hanein, S.; Fares-Taie, L.; Gerber, S.; Defoort-Dhellemmes, S.; Vojtek, A.M.; et al. Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis with CEP290 mutations. J. Med. Genet. 2010, 47, 829–834. [Google Scholar] [CrossRef]
- Drivas, T.G.; Wojno, A.P.; Tucker, B.A.; Stone, E.M.; Bennett, J. Basal exon skipping and genetic pleiotropy: A predictive model of disease pathogenesis. Sci. Trans. Med. 2015, 7, 291ra97. [Google Scholar] [CrossRef]
- Barny, I.; Perrault, I.; Michel, C.; Soussan, M.; Goudin, N.; Rio, M.; Thomas, S.; Attié-Bitach, T.; Hamel, C.; Dollfus, H.; et al. Basal exon skipping and nonsense-associated altered splicing allows bypassing complete CEP290 loss-of-function in individuals with unusually mild retinal disease. Hum. Mol. Genet. 2018, 15, 2689–2702. [Google Scholar] [CrossRef]
- Littink, K.W.; Pott, J.W.; Collin, R.W.; Kroes, H.Y.; Verheij, J.B.; Blokland, E.A.; de Castro Miro, M.; Hoyng, C.B.; Klaver, C.C.; Joenekoop, R.K.; et al. A novel nonsense mutation in CEP290 induces exon skipping and leads to a relatively mild retinal phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3646–3652. [Google Scholar] [CrossRef]
- Roosing, S.; Cremers, F.P.M.; Riemslag, F.C.C.; Zonneveld-Vrieling, M.N.; Talsma, H.E.; Klessens-Godfroy, F.J.M.; den Hollander, A.I.; van den Born, L.I. A rare form of retinal dystrophy caused by hypomorphic nonsense mutations in CEP290. Genes 2017, 8, 208. [Google Scholar] [CrossRef]
- Disterer, P.; Kryczka, A.; Liu, Y.; Badi, Y.E.; Wong, J.J.; Owen, J.S.; Khoo, B. Development of therapeutic splice-switching oligonucleotides. Hum. Gene Ther. 2014, 25, 587–598. [Google Scholar] [CrossRef]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peränen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. CP110 Suppresses primary cilia formation through its interaction with cep290, a protein deficient in human ciliary disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef]
- Kim, J.; Krishnaswami, S.R.; Gleeson, J.G. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum. Mol. Genet. 2008, 17, 3796–3805. [Google Scholar] [CrossRef]
- Stowe, T.R.; Wilkinson, C.J.; Iqbal, A.; Stearns, T. The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol. Biol. Cell 2012, 23, 3322–3335. [Google Scholar] [CrossRef]
- Ramsbottom, S.A.; Molinari, E.; Srivastava, S.; Silberman, F.; Henry, C.; Alkanderi, S.; Devlin, L.A.; White, K.; Steel, D.H.; Saunier, S.; et al. Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model. Proc. Natl. Acad. Sci. USA 2018, 115, 12489–12494. [Google Scholar] [CrossRef]
- Drivas, T.G.; Holzbaur, E.L.F.; Bennett, J. Disruption of CEP290 microtubule/membrane-binding domains causes retinal degeneration. J. Clin. Investig. 2013, 123, 4525–4539. [Google Scholar] [CrossRef]
- Rachel, R.A.; Yamamoto, E.A.; Dewanjee, M.K.; May-Simera, H.L.; Sergeev, Y.V.; Hackett, A.N.; Pohida, K.; Munasinghe, J.; Gotoh, N.; Wickstead, B.; et al. CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum. Mol. Genet. 2015, 24, 3775–3791. [Google Scholar] [CrossRef]
- Chang, B.; Khanna, H.; Hawes, N.; Jimeno, D.; He, S.; Lillo, C.; Parapuram, S.K.; Cheng, H.; Scott, A.; Hurd, R.E.; et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 2006, 15, 1847–1857. [Google Scholar] [CrossRef]
- Shimada, H.; Lu, Q.; Insinna-Kettenhofen, C.; Nagashima, K.; English, M.A.; Semler, E.M.; Mahgerefteh, J.; Cideciyan, A.V.; Li, T.; Brooks, B.P.; et al. In Vitro modeling using ciliopathy-patient-derived cells reveals distinct cilia dysfunctions caused by CEP290 mutations. Cell Rep. 2017, 20, 384–396. [Google Scholar] [CrossRef]
- Barny, I.; Perrault, I.; Rio, M.; Dollfus, H.; Defoort-Dhellemmes, S.; Kaplan, J.; Rozet, J.-M.; Gerard, X. Description of two siblings with apparently severe CEP290 mutations and unusually mild retinal disease unrelated to basal exon skipping or nonsense-associated altered splicing. Adv. Exp. Med. Biol. (under review).
- Valentine, C.R. The association of nonsense codons with exon skipping. Mutat. Res./Rev. Mutat. Res. 1998, 411, 87–117. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285. [Google Scholar] [CrossRef] [PubMed]
- Shiga, N.; Takeshima, Y.; Sakamoto, H.; Inoue, K.; Yokota, Y.; Yokoyama, M.; Matsuo, M. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J. Clin. Investig. 1997, 100, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; Baere, E.D. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef]
- Mokrzan, E.M.; Lewis, J.S.; Mykytyn, K. Differences in renal tubule primary cilia length in a mouse model of bardet-biedl syndrome. NEE 2007, 106, e88–e96. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.A.; Bukanov, N.O.; Husson, H.; Russo, R.J.; Barry, T.C.; Taylor, A.L.; Beier, D.R.; Ibraghimov-Beskrovnaya, O. Development of Polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. JASN 2006, 17, 2821–2831. [Google Scholar] [CrossRef]
- Sohara, E.; Luo, Y.; Zhang, J.; Manning, D.K.; Beier, D.R.; Zhou, J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J. Am. Soc. Nephrol. 2008, 19, 469–476. [Google Scholar] [CrossRef]
- Tammachote, R.; Hommerding, C.J.; Sinders, R.M.; Miller, C.A.; Czarnecki, P.G.; Leightner, A.C.; Salisbury, J.L.; Ward, C.J.; Torres, V.E.; Gattone, V.H., 2nd; et al. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum. Mol. Genet. 2009, 18, 3311–3323. [Google Scholar] [CrossRef]
- He, M.; Subramanian, R.; Bangs, F.; Omelchenko, T.; Liem, K.F. Jr.; Kapoor, T.M.; Anderson, K.V. The kinesin-4 protein KIF7 regulates mammalian hedgehog signaling by organizing the cilia tip compartment. Nat. Cell. Biol. 2014, 16, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.A.; de Vrieze, E.; Alazami, A.M.; Alzahrani, F.; Malarkey, E.B.; Sorush, N.; Tebbe, L.; Kuhns, S.; van Dam, T.J.; Alhashem, A.; et al. KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol. 2015, 16, 293. [Google Scholar] [CrossRef] [PubMed]
- Stayner, C.; Poole, C.A.; McGlashan, S.R.; Pilanthananond, M.; Brauning, R.; Markie, D.; Lett, B.; Slobbe, L.; Chae, A.; Johnstone, A.C.; et al. An ovine hepatorenal fibrocystic model of a Meckel-like syndrome associated with dysmorphic primary cilia and TMEM67 mutations. Sci. Rep. 2017, 7, 1601. [Google Scholar] [CrossRef]
- Nikopoulos, K.; Farinelli, P.; Giangreco, B.; Tsika, C.; Royer-Bertand, B.; Mbefo, M.K.; Bedoni, N.; Kjellström, U.; El Zaoui, I.; Di Gioia, S.A.; et al. Mutations in CEP78 cause cone-rod dystrophy and hearing loss associated with primary-cilia defects. Am. J. Hum. Genet. 2016, 99, 770–776. [Google Scholar] [CrossRef]
- Srivastava, S.; Ramsbottom, S.A.; Molinari, E.; Alkanderi, S.; Filby, A.; White, K.; Henry, C.; Saunier, S.; Miles, C.G.; Sayer, J.A. A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum. Mol. Genet. 2017, 26, 4657–4667. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; van Beersum, S.E.C.; Peters, T.A.; Roepman, R.; Cremers, F.P.M.; Collin, R.W.J. Unexpected CEP290 mRNA splicing in a humanized knock-in mouse model for leber congenital amaurosis. PLoS ONE 2013, 8, e79369. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.F.; Qin, H.; Brenni, M.R.; Rosenbaum, J.L. Flagellar length control system: Testing a simple model based on intraflagellar transport and turnover. Mol. Biol. Cell 2005, 16, 270–278. [Google Scholar] [CrossRef]
- Tanackovic, G.; Rivolta, C. PRPF31 alternative splicing and expression in human retina. Ophthalmic Genet. 2009, 30, 76–83. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide | EX-SKIP Predictions | HOT-SKIP Predictions | Skipping Predictions of Mutant Allele Compared to WT Allele | |||||
|---|---|---|---|---|---|---|---|---|
| ESS | ESE | ESS/ESE | ESS | ESE | ESS/ESE | |||
| c.4723 (exon 36) | A | 12 | 88 | 0.14 | 2 | 17 | 0.12 | - |
| T | 19 | 75 | 0.25 | 9 | 4 | 2.25 | Higher chance | |
| G | 13 | 87 | 0.15 | 3 | 16 | 0.19 | Higher chance | |
| C | 11 | 82 | 0.13 | 1 | 11 | 0.09 | Lower chance | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barny, I.; Perrault, I.; Michel, C.; Goudin, N.; Defoort-Dhellemmes, S.; Ghazi, I.; Kaplan, J.; Rozet, J.-M.; Gerard, X. AON-Mediated Exon Skipping to Bypass Protein Truncation in Retinal Dystrophies Due to the Recurrent CEP290 c.4723A > T Mutation. Fact or Fiction? Genes 2019, 10, 368. https://doi.org/10.3390/genes10050368
Barny I, Perrault I, Michel C, Goudin N, Defoort-Dhellemmes S, Ghazi I, Kaplan J, Rozet J-M, Gerard X. AON-Mediated Exon Skipping to Bypass Protein Truncation in Retinal Dystrophies Due to the Recurrent CEP290 c.4723A > T Mutation. Fact or Fiction? Genes. 2019; 10(5):368. https://doi.org/10.3390/genes10050368
Chicago/Turabian StyleBarny, Iris, Isabelle Perrault, Christel Michel, Nicolas Goudin, Sabine Defoort-Dhellemmes, Imad Ghazi, Josseline Kaplan, Jean-Michel Rozet, and Xavier Gerard. 2019. "AON-Mediated Exon Skipping to Bypass Protein Truncation in Retinal Dystrophies Due to the Recurrent CEP290 c.4723A > T Mutation. Fact or Fiction?" Genes 10, no. 5: 368. https://doi.org/10.3390/genes10050368
APA StyleBarny, I., Perrault, I., Michel, C., Goudin, N., Defoort-Dhellemmes, S., Ghazi, I., Kaplan, J., Rozet, J.-M., & Gerard, X. (2019). AON-Mediated Exon Skipping to Bypass Protein Truncation in Retinal Dystrophies Due to the Recurrent CEP290 c.4723A > T Mutation. Fact or Fiction? Genes, 10(5), 368. https://doi.org/10.3390/genes10050368