Functional Proteomics of Nuclear Proteins in Tetrahymena thermophila: A Review

 ,
, {kind=link}
{kind=link}
Abstract
:1. Expression, Functional and Comparative Proteomics
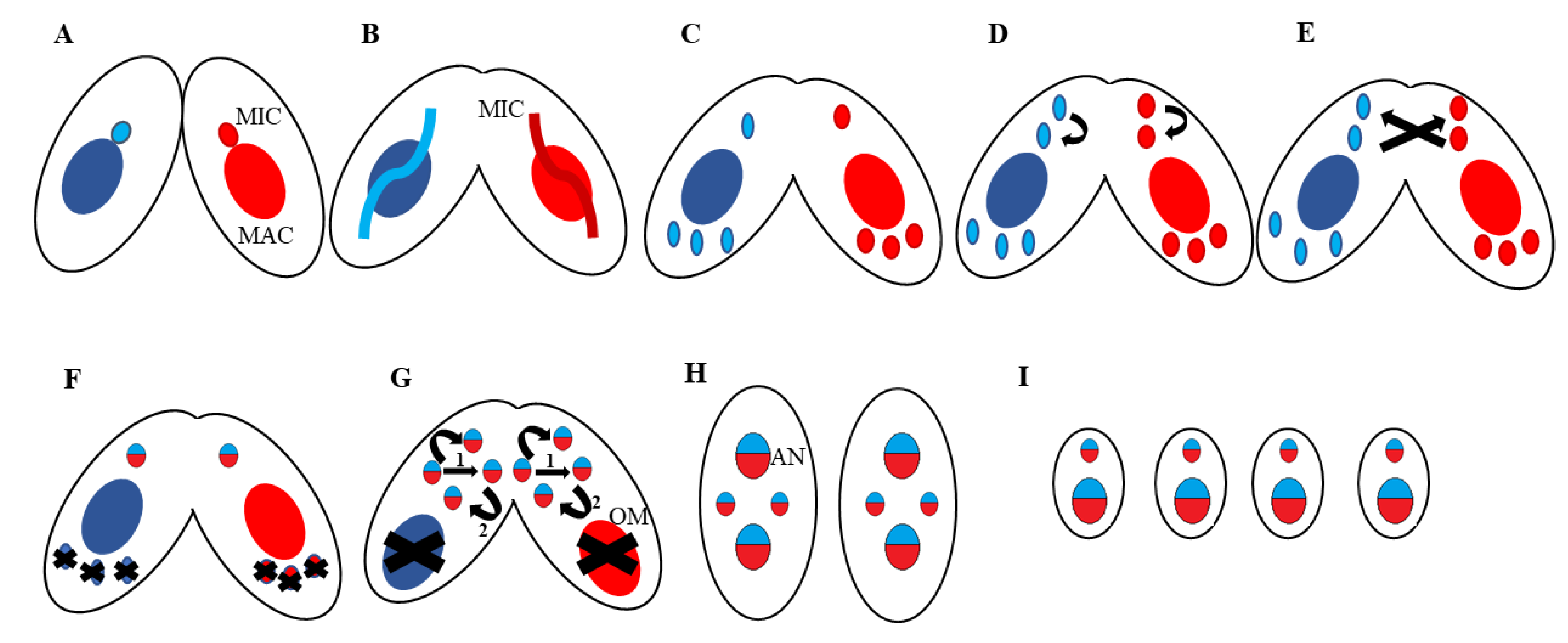
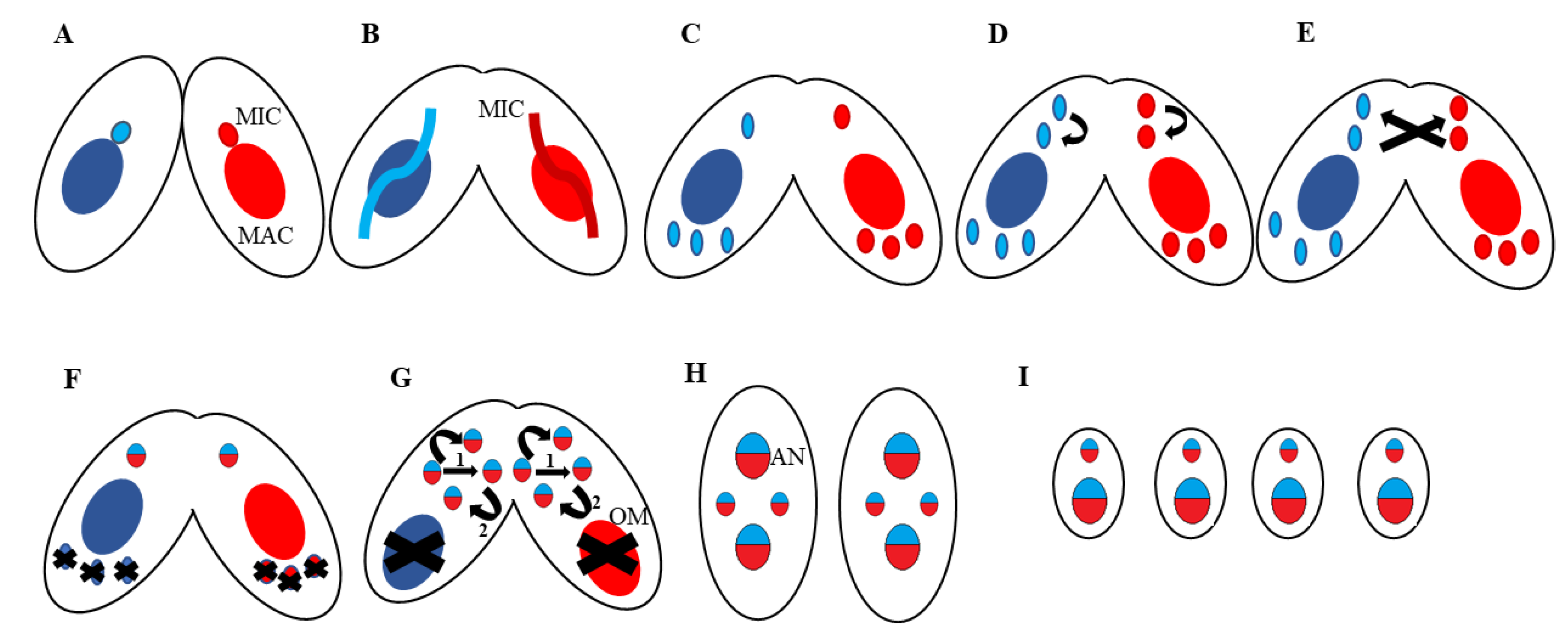
2. Tetrahymena thermophila: A Useful Proteomic Model for Nuclear Events
3. Identification of Core Histones and their Variants
4. Telomeres and Telomerase
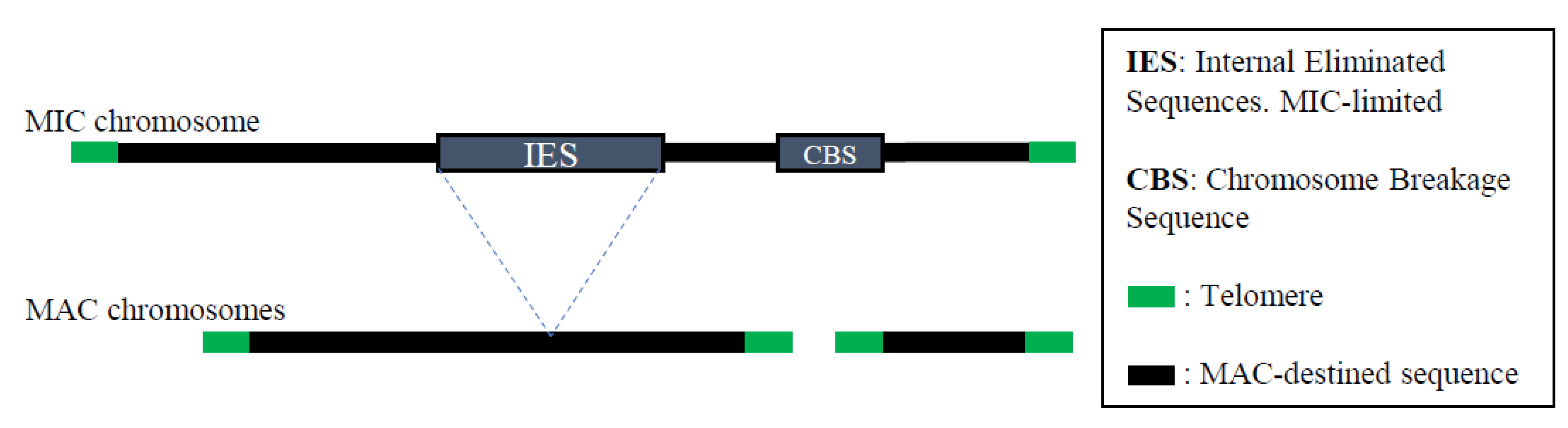
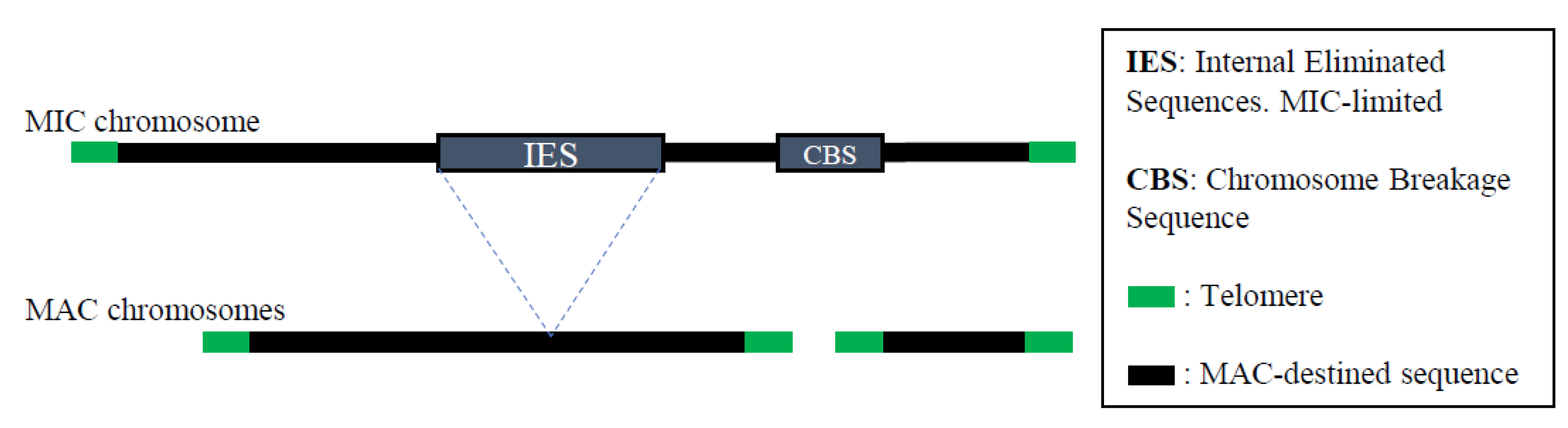
5. Programmed DNA Rearrangements
6. RNAi Mechanisms
7. Epigenetics
8. Chromatin Assembly
9. Chromatin Remodeling
10. Transcription
11. Comparative Proteomics
12. New Proteomic Technologies for the Characterization of Tetrahymena Nuclear Proteins
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- James, P. Protein identification in the post-genome era: The rapid rise of proteomics. Q. Rev. Biophys. 1997, 30, 279–331. [Google Scholar] [CrossRef] [PubMed]
- Köcher, T.; Superti-Furga, G. Mass spectrometry-based functional proteomics: From molecular machines to protein networks. Nat. Methods 2007, 4, 807–815. [Google Scholar] [CrossRef]
- Rost, B. Did evolution leap to create the protein universe? Curr. Opin. Struct. Biol. 2002, 12, 409–416. [Google Scholar] [CrossRef]
- Alberts, B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 1998, 92, 291–294. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Owen, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Saettone, A.; Garg, J.; Lambert, J.-P.; Nabeel-Shah, S.; Ponce, M.; Burtch, A.; Thuppu Mudalige, C.; Gingras, A.-C.; Pearlman, R.E.; Fillingham, J. The bromodomain-containing protein Ibd1 links multiple chromatin-related protein complexes to highly expressed genes in Tetrahymena thermophila. Epigenetics Chromatin 2018, 11, 10. [Google Scholar] [CrossRef]
- Ashraf, K.; Nabeel-Shah, S.; Garg, J.; Saettone, A.; Derynck, J.; Gingras, A.-C.; Lambert, J.-P.; Pearlman, R.E.; Fillingham, J. Proteomic analysis of histones H2A/H2B and variant Hv1 in Tetrahymena thermophila reveals an ancient network of chaperones. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef]
- Butland, G.; Peregrín-Alvarez, J.M.; Li, J.; Yang, W.; Yang, X.; Canadien, V.; Starostine, A.; Richards, D.; Beattie, B.; Krogan, N.; et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 2005, 433, 531–537. [Google Scholar] [CrossRef]
- Krogan, N.J.; Cagney, G.; Yu, H.; Zhong, G.; Guo, X.; Ignatchenko, A.; Li, J.; Pu, S.; Datta, N.; Tikuisis, A.P.; et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440, 637–643. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Sharan, R.; Suthram, S.; Kelley, R.M.; Kuhn, T.; McCuine, S.; Uetz, P.; Sittler, T.; Karp, R.M.; Ideker, T. Conserved patterns of protein interaction in multiple species. Proc. Natl. Acad. Sci. USA 2005, 102, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef]
- Hu, P.; Janga, S.C.; Babu, M.; Díaz-Mejía, J.J.; Butland, G.; Yang, W.; Pogoutse, O.; Guo, X.; Phanse, S.; Wong, P.; et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009, 7, e96. [Google Scholar] [CrossRef]
- Guruharsha, K.G.; Rual, J.-F.; Zhai, B.; Mintseris, J.; Vaidya, P.; Vaidya, N.; Beekman, C.; Wong, C.; Rhee, D.Y.; Cenaj, O.; et al. A protein complex network of Drosophila melanogaster. Cell 2011, 147, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Simonis, N.; Rual, J.-F.; Carvunis, A.-R.; Tasan, M.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Sahalie, J.M.; Venkatesan, K.; Gebreab, F.; et al. Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat. Methods 2009, 6, 47–54. [Google Scholar] [CrossRef]
- Orias, E.; Cervantes, M.D.; Hamilton, E.P. Tetrahymena thermophila, a unicellular eukaryote with separate germline and somatic genomes. Res. Microbiol. 2011, 162, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Allis, C.D. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc. Natl. Acad. Sci. USA 1995, 92, 6364–6368. [Google Scholar] [CrossRef]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Chalker, D.L.; Yao, M.-C. DNA elimination in ciliates: Transposon domestication and genome surveillance. Annu. Rev. Genet. 2011, 45, 227–246. [Google Scholar] [CrossRef]
- Streit, A.; Wang, J.; Kang, Y.; Davis, R.E. Gene silencing and sex determination by programmed DNA elimination in parasitic nematodes. Curr. Opin. Microbiol. 2016, 32, 120–127. [Google Scholar] [CrossRef]
- Ruehle, M.D.; Orias, E.; Pearson, C.G. Tetrahymena as a Unicellular Model Eukaryote: Genetic and Genomic Tools. Genetics 2016, 203, 649–665. [Google Scholar] [CrossRef]
- Eisen, J.A.; Coyne, R.S.; Wu, M.; Wu, D.; Thiagarajan, M.; Wortman, J.R.; Badger, J.H.; Ren, Q.; Amedeo, P.; Jones, K.M.; et al. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol. 2006, 4, e286. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Kapusta, A.; Huvos, P.E.; Bidwell, S.L.; Zafar, N.; Tang, H.; Hadjithomas, M.; Krishnakumar, V.; Badger, J.H.; Caler, E.V.; et al. Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. Elife 2016, 5, e19090. [Google Scholar] [CrossRef]
- Chalker, D.L. Transformation and Strain Engineering of Tetrahymena. Methods Cell Biol. 2012, 109, 327–345. [Google Scholar]
- Akematsu, T.; Fukuda, Y.; Garg, J.; Fillingham, J.S.; Pearlman, R.E.; Loidl, J. Post-meiotic DNA double-strand breaks occur in Tetrahymena, and require Topoisomerase II and Spo11. Elife 2017, 6, e26176. [Google Scholar] [CrossRef]
- Akematsu, T.; Findlay, A.; Fukuda, Y.; Pearlman, R.E.; Loidl, J.; Orias, E.; P Hamilton, E. Resistance to 6-Methylpurine is Conferred by Defective Adenine Phosphoribosyltransferase in Tetrahymena. Genes 2018, 9, 179. [Google Scholar] [CrossRef]
- Stover, N.A.; Krieger, C.J.; Binkley, G.; Dong, Q.; Fisk, D.G.; Nash, R.; Sethuraman, A.; Weng, S.; Cherry, J.M. Tetrahymena Genome Database (TGD): A new genomic resource for Tetrahymena thermophila research. Nucleic Acids Res. 2006, 34, D500–D503. [Google Scholar] [CrossRef]
- Stover, N.A.; Punia, R.S.; Bowen, M.S.; Dolins, S.B.; Clark, T.G. Tetrahymena Genome Database Wiki: A community-maintained model organism database. Database 2012, 2012, bas007. [Google Scholar] [CrossRef]
- Xiong, J.; Lu, X.; Zhou, Z.; Chang, Y.; Yuan, D.; Tian, M.; Zhou, Z.; Wang, L.; Fu, C.; Orias, E.; et al. Transcriptome analysis of the model protozoan, tetrahymena thermophila, using deep RNA sequencing. PLoS ONE 2012, 7, 1–13. [Google Scholar] [CrossRef]
- Xiong, J.; Lu, Y.; Feng, J.; Yuan, D.; Tian, M.; Chang, Y.; Fu, C.; Wang, G.; Zeng, H.; Miao, W. Tetrahymena functional genomics database (TetraFGD): An integrated resource for Tetrahymena functional genomics. Database 2013, 2013, bat008. [Google Scholar] [CrossRef]
- Gorovsky, M.A. Studies on nuclear structure and function in Tetrahymena pyriformis. II. Isolation of macro- and micronuclei. J. Cell Biol. 1970, 47, 619–630. [Google Scholar] [CrossRef]
- Gorovsky, M.A. Studies on nuclear structure and function in Tetrahymena pyriformis. 3. Comparison of the histones of macro- and micronuclei by quantitative polyacrylamide gel electrophoresis. J. Cell Biol. 1970, 47, 631–636. [Google Scholar] [CrossRef]
- Allis, C.D.; Glover, C.V.; Gorovsky, M.A. Micronuclei of Tetrahymena contain two types of histone H3. Proc. Natl. Acad. Sci. USA 1979, 76, 4857–4861. [Google Scholar] [CrossRef]
- Allis, C.D.; Bowen, J.K.; Abraham, G.N.; Glover, C.V.C.; Gorovsky, M.A. Proteolytic processing of histone H3 in chromatin: A physiologically regulated event in Tetrahymena micronuclei. Cell 1980, 20, 55–64. [Google Scholar] [CrossRef]
- Allis, C.D.; Glover, C.V.C.; Bowen, J.K.; Gorovsky, M.A. Histone variants specific to the transcriptionally active, amitotically dividing macronucleus of the unicellular eucaryote, Tetrahymena thermophila. Cell 1980, 20, 609–617. [Google Scholar] [CrossRef]
- Schulman, I.G.; Cook, R.G.; Richman, R.; Allis, C.D. Tetrahymena contain two distinct and unusual high mobility group (HMG)-like proteins. J. Cell Biol. 1987, 104, 1485–1494. [Google Scholar] [CrossRef]
- Allis, C.D.; Allen, R.L.; Wiggins, J.C.; Chicoine, L.G.; Richman, R. Proteolytic processing of h1-like histones in chromatin: A physiologically and developmentally regulated event in Tetrahymena micronuclei. J. Cell Biol. 1984, 99, 1669–1677. [Google Scholar] [CrossRef]
- Huang, H.; Wiley, E.A.; Lending, C.R.; Allis, C.D. An HP1-like protein is missing from transcriptionally silent micronuclei of Tetrahymena. Proc. Natl. Acad. Sci. USA 1998, 95, 13624–13629. [Google Scholar] [CrossRef]
- Blackburn, E.H. The molecular structure of centromeres and telomeres. Annu. Rev. Biochem. 1984, 53, 163–194. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987, 51, 887–898. [Google Scholar] [CrossRef]
- Collins, K.; Kobayashi, R.; Greider, C.W. Purification of tetrahymena telomerase and cloning of genes encoding the two protein components of the enzyme. Cell 1995, 81, 677–686. [Google Scholar] [CrossRef]
- Nakamura, T.M. Telomerase Catalytic Subunit Homologs from Fission Yeast and Human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef]
- Collins, K.; Gandhi, L. The reverse transcriptase component of the Tetrahymena telomerase ribonucleoprotein complex. Proc. Natl. Acad. Sci. USA 1998, 95, 8485–8490. [Google Scholar] [CrossRef]
- Rigaut, G.; Shevchenko, A.; Rutz, B.; Wilm, M.; Mann, M.; Séraphin, B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999, 17, 1030–1032. [Google Scholar] [CrossRef]
- Witkin, K.L.; Collins, K. Holoenzyme proteins required for the physiological assembly and activity of telomerase. Genes Dev. 2004, 18, 1107–1118. [Google Scholar] [CrossRef]
- Gaertig, J.; Gao, Y.; Tishgarten, T.; Clark, T.G.; Dickerson, H.W. Surface display of a parasite antigen in the ciliate Tetrahymena thermophila. Nat. Biotechnol. 1999, 17, 462–465. [Google Scholar] [CrossRef]
- Prathapam, R.; Witkin, K.L.; O’Connor, C.M.; Collins, K. A telomerase holoenzyme protein enhances telomerase RNA assembly with telomerase reverse transcriptase. Nat. Struct. Mol. Biol. 2005, 12, 252–257. [Google Scholar] [CrossRef]
- Witkin, K.L.; Prathapam, R.; Collins, K. Positive and negative regulation of Tetrahymena telomerase holoenzyme. Mol. Cell. Biol. 2007, 27, 2074–2083. [Google Scholar] [CrossRef]
- Min, B.; Collins, K. An RPA-Related Sequence-Specific DNA-Binding Subunit of Telomerase Holoenzyme Is Required for Elongation Processivity and Telomere Maintenance. Mol. Cell 2009, 36, 609–619. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, H.; Cash, D.D.; Miracco, E.J.; Loo, R.R.O.; Upton, H.E.; Cascio, D.; Johnson, R.O.B.; Collins, K.; Loo, J.A.; et al. Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science 2015, 350, aab4070. [Google Scholar] [CrossRef]
- Upton, H.E.; Chan, H.; Feigon, J.; Collins, K. Shared Subunits of Tetrahymena Telomerase Holoenzyme and Replication Protein A Have Different Functions in Different Cellular Complexes. J. Biol. Chem. 2017, 292, 217–228. [Google Scholar] [CrossRef]
- Jacob, N.K.; Lescasse, R.; Linger, B.R.; Price, C.M. Tetrahymena POT1a regulates telomere length and prevents activation of a cell cycle checkpoint. Mol. Cell. Biol. 2007, 27, 1592–1601. [Google Scholar] [CrossRef]
- Linger, B.R.; Morin, G.B.; Price, C.M. The Pot1a-associated proteins Tpt1 and Pat1 coordinate telomere protection and length regulation in Tetrahymena. Mol. Biol. Cell 2011, 22, 4161–4170. [Google Scholar] [CrossRef]
- Premkumar, V.L.; Cranert, S.; Linger, B.R.; Morin, G.B.; Minium, S.; Price, C. The 3′ overhangs at Tetrahymena thermophila telomeres are packaged by four proteins, Pot1a, Tpt1, Pat1, and Pat2. Eukaryot. Cell 2014, 13, 240–245. [Google Scholar] [CrossRef]
- Yao, M.-C.; Chao, J.-L.; Cheng, C.-Y. Programmed Genome Rearrangements in Tetrahymena. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Madireddi, M.T.; Davis, M.C.; Allis, C.D. Identification of a novel polypeptide involved in the formation of DNA-containing vesicles during macronuclear development in Tetrahymena. Dev. Biol. 1994, 165, 418–431. [Google Scholar] [CrossRef]
- Madireddi, M.T.; Coyne, R.S.; Smothers, J.F.; Mickey, K.M.; Yao, M.-C.; Allis, C.D. Pdd1p, A Novel Chromodomain-Containing Protein, Links Heterochromatin Assembly and DNA Elimination in Tetrahymena. Cell 1996, 87, 75–84. [Google Scholar] [CrossRef]
- Smothers, J.F.; Mizzen, C.A.; Tubbert, M.M.; Cook, R.G.; Allis, C.D. Pdd1p associates with germline-restricted chromatin and a second novel anlagen-enriched protein in developmentally programmed DNA elimination structures. Development 1997, 124, 4537–4545. [Google Scholar]
- Nikiforov, M.A.; Gorovsky, M.A.; Allis, C.D. A novel chromodomain protein, pdd3p, associates with internal eliminated sequences during macronuclear development in Tetrahymena thermophila. Mol. Cell. Biol. 2000, 20, 4128–4134. [Google Scholar] [CrossRef]
- Mochizuki, K.; Fine, N.A.; Fujisawa, T.; Gorovsky, M.A. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in Tetrahymena. Cell 2002, 110, 689–699. [Google Scholar] [CrossRef]
- Taverna, S.D.; Coyne, R.S.; Allis, C.D. Methylation of histone h3 at lysine 9 targets programmed DNA elimination in tetrahymena. Cell 2002, 110, 701–711. [Google Scholar] [CrossRef]
- Allis, C.D.; Dennison, D.K. Identification and purification of young macronuclear anlagen from conjugating cells of Tetrahymena thermophila. Dev. Biol. 1982, 93, 519–533. [Google Scholar] [CrossRef]
- DeSouza, L.V.; Voisin, S.N.; Siu, K.W.M. iTRAQ-Labeling for Biomarker Discovery. Methods Mol. Biol. 2013, 1002, 105–114. [Google Scholar]
- Chalker, D.L.; Yao, M.C. Non-Mendelian, heritable blocks to DNA rearrangement are induced by loading the somatic nucleus of Tetrahymena thermophila with germ line-limited DNA. Mol. Cell. Biol. 1996, 16, 3658–3667. [Google Scholar] [CrossRef]
- Aronica, L.; Bednenko, J.; Noto, T.; DeSouza, L.V.; Siu, K.W.M.; Loidl, J.; Pearlman, R.E.; Gorovsky, M.A.; Mochizuki, K. Study of an RNA helicase implicates small RNA-noncoding RNA interactions in programmed DNA elimination in Tetrahymena. Genes Dev. 2008, 22, 2228–2241. [Google Scholar] [CrossRef]
- Bednenko, J.; Noto, T.; DeSouza, L.V.; Siu, K.W.M.; Pearlman, R.E.; Mochizuki, K.; Gorovsky, M.A. Two GW Repeat Proteins Interact with Tetrahymena thermophila Argonaute and Promote Genome Rearrangement. Mol. Cell. Biol. 2009, 29, 5020–5030. [Google Scholar] [CrossRef]
- Martindale, D.W.; Bruns, P.J. Cloning of abundant mRNA species present during conjugation of Tetrahymena thermophila: Identification of mRNA species present exclusively during meiosis. Mol. Cell. Biol. 1983, 3, 1857–1865. [Google Scholar] [CrossRef]
- Noto, T.; Kurth, H.M.; Kataoka, K.; Aronica, L.; DeSouza, L.V.; Siu, K.W.M.; Pearlman, R.E.; Gorovsky, M.A.; Mochizuki, K. The Tetrahymena argonaute-binding protein Giw1p directs a mature argonaute-siRNA complex to the nucleus. Cell 2010, 140, 692–703. [Google Scholar] [CrossRef]
- Lee, S.R.; Collins, K. Two classes of endogenous small RNAs in Tetrahymena thermophila. Genes Dev. 2006, 20, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Collins, K. Physical and functional coupling of RNA-dependent RNA polymerase and Dicer in the biogenesis of endogenous siRNAs. Nat. Struct. Mol. Biol. 2007, 14, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Talsky, K.B.; Collins, K. A single RNA-dependent RNA polymerase assembles with mutually exclusive nucleotidyl transferase subunits to direct different pathways of small RNA biogenesis. RNA 2009, 15, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Talsky, K.B.; Collins, K. Strand-asymmetric endogenous Tetrahymena small RNA production requires a previously uncharacterized uridylyltransferase protein partner. RNA 2012, 18, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Couvillion, M.T.; Lee, S.R.; Hogstad, B.; Malone, C.D.; Tonkin, L.A.; Sachidanandam, R.; Hannon, G.J.; Collins, K. Sequence, biogenesis, and function of diverse small RNA classes bound to the Piwi family proteins of Tetrahymena thermophila. Genes Dev. 2009, 23, 2016–2032. [Google Scholar] [CrossRef] [PubMed]
- Couvillion, M.T.; Sachidanandam, R.; Collins, K. A growth-essential Tetrahymena Piwi protein carries tRNA fragment cargo. Genes Dev. 2010, 24, 2742–2747. [Google Scholar] [CrossRef] [PubMed]
- Couvillion, M.T.; Bounova, G.; Purdom, E.; Speed, T.P.; Collins, K. A Tetrahymena Piwi bound to mature tRNA 3′ fragments activates the exonuclease Xrn2 for RNA processing in the nucleus. Mol. Cell 2012, 48, 509–520. [Google Scholar] [CrossRef]
- Allis, C.D.; Chicoine, L.G.; Richman, R.; Schulman, I.G. Deposition-related histone acetylation in micronuclei of conjugating Tetrahymena. Proc. Natl. Acad. Sci. USA 1985, 82, 8048–8052. [Google Scholar] [CrossRef]
- Chicoine, L.G.; Schulman, I.G.; Richman, R.; Cook, R.G.; Allis, C.D. Nonrandom utilization of acetylation sites in histones isolated from Tetrahymena. Evidence for functionally distinct H4 acetylation sites. J. Biol. Chem. 1986, 261, 1071–1076. [Google Scholar] [PubMed]
- Sobel, R.E.; Cook, R.G.; Perry, C.A.; Annunziato, A.T.; Allis, C.D. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc. Natl. Acad. Sci. USA 1995, 92, 1237–1241. [Google Scholar] [CrossRef]
- Mizzen, C.A.; Dou, Y.; Liu, Y.; Cook, R.G.; Gorovsky, M.A.; Allis, C.D. Identification and mutation of phosphorylation sites in a linker histone. Phosphorylation of macronuclear H1 is not essential for viability in tetrahymena. J. Biol. Chem. 1999, 274, 14533–14536. [Google Scholar] [CrossRef]
- Dou, Y.; Mizzen, C.A.; Abrams, M.; Allis, C.D.; Gorovsky, M.A. Phosphorylation of linker histone H1 regulates gene expression in vivo by mimicking H1 removal. Mol. Cell 1999, 4, 641–647. [Google Scholar] [CrossRef]
- Dou, Y.; Song, X.; Liu, Y.; Gorovsky, M.A. The H1 phosphorylation state regulates expression of CDC2 and other genes in response to starvation in Tetrahymena thermophila. Mol. Cell. Biol. 2005, 25, 3914–3922. [Google Scholar] [CrossRef]
- Strahl, B.D.; Ohba, R.; Cook, R.G.; Allis, C.D. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl. Acad. Sci. USA 1999, 96, 14967–14972. [Google Scholar] [CrossRef]
- Garcia, B.A.; Hake, S.B.; Diaz, R.L.; Kauer, M.; Morris, S.A.; Recht, J.; Shabanowitz, J.; Mishra, N.; Strahl, B.D.; Allis, C.D.; et al. Organismal differences in post-translational modifications in histones H3 and H4. J. Biol. Chem. 2007, 282, 7641–7655. [Google Scholar] [CrossRef]
- Masumoto, H.; Hawke, D.; Kobayashi, R.; Verreault, A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 2005, 436, 294–298. [Google Scholar] [CrossRef]
- Recht, J.; Tsubota, T.; Tanny, J.C.; Diaz, R.L.; Berger, J.M.; Zhang, X.; Garcia, B.A.; Shabanowitz, J.; Burlingame, A.L.; Hunt, D.F.; et al. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc. Natl. Acad. Sci. USA 2006, 103, 6988–6993. [Google Scholar] [CrossRef]
- Liu, Y.; Taverna, S.D.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F.; Allis, C.D. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 2007, 21, 1530–1545. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Taverna, S.D.; Ueberheide, B.M.; Liu, Y.; Tackett, A.J.; Diaz, R.L.; Shabanowitz, J.; Chait, B.T.; Hunt, D.F.; Allis, C.D. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc. Natl. Acad. Sci. USA 2007, 104, 2086–2091. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, S.; Molascon, A.J.; Wang, Z.; Gorovsky, M.A.; Liu, Y.; Andrews, P.C. Bioinformatic and proteomic analysis of bulk histones reveals PTM crosstalk and chromatin features. J. Proteome Res. 2014, 13, 3330–3337. [Google Scholar] [CrossRef]
- Papazyan, R.; Voronina, E.; Chapman, J.R.; Luperchio, T.R.; Gilbert, T.M.; Meier, E.; Mackintosh, S.G.; Shabanowitz, J.; Tackett, A.J.; Reddy, K.L.; et al. Methylation of histone H3K23 blocks DNA damage in pericentric heterochromatin during meiosis. Elife 2014, 3, e02996. [Google Scholar] [CrossRef]
- Campos, E.I.; Fillingham, J.; Li, G.; Zheng, H.; Voigt, P.; Kuo, W.-H.W.; Seepany, H.; Gao, Z.; Day, L.A.; Greenblatt, J.F.; et al. The program for processing newly synthesized histones H3.1 and H4. Nat. Struct. Mol. Biol. 2010, 17, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Krogan, N.J.; Keogh, M.-C.; Datta, N.; Sawa, C.; Ryan, O.W.; Ding, H.; Haw, R.A.; Pootoolal, J.; Tong, A.; Canadien, V.; et al. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol. Cell 2003, 12, 1565–1576. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- Campos, E.I.; Smits, A.H.; Kang, Y.-H.; Landry, S.; Escobar, T.M.; Nayak, S.; Ueberheide, B.M.; Durocher, D.; Vermeulen, M.; Hurwitz, J.; et al. Analysis of the Histone H3.1 Interactome: A Suitable Chaperone for the Right Event. Mol. Cell 2015, 60, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Mendiratta, S.; Gatto, A.; Almouzni, G. Histone supply: Multitiered regulation ensures chromatin dynamics throughout the cell cycle. J. Cell Biol. 2019, 218, 39–54. [Google Scholar] [CrossRef] [PubMed]
- De Koning, L.; Corpet, A.; Haber, J.E.; Almouzni, G. Histone chaperones: An escort network regulating histone traffic. Nat. Struct. Mol. Biol. 2007, 14, 997–1007. [Google Scholar] [CrossRef]
- Garg, J.; Lambert, J.-P.; Karsou, A.; Marquez, S.; Nabeel-Shah, S.; Bertucci, V.; Retnasothie, D.V.; Radovani, E.; Pawson, T.; Gingras, A.-C.; et al. Conserved Asf1-importin β physical interaction in growth and sexual development in the ciliate Tetrahymena thermophila. J. Proteom. 2013, 94, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Larsen, B.; Lin, Z.-Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.-C.; Nesvizhskii, A.I. SAINT: Probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef]
- Choi, H.; Liu, G.; Mellacheruvu, D.; Tyers, M.; Gingras, A.-C.; Nesvizhskii, A.I. Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr. Protoc. Bioinform. 2012. [Google Scholar] [CrossRef]
- Miao, W.; Xiong, J.; Bowen, J.; Wang, W.; Liu, Y.; Braguinets, O.; Grigull, J.; Pearlman, R.E.; Orias, E.; Gorovsky, M.A. Microarray analyses of gene expression during the Tetrahymena thermophila life cycle. PLoS ONE 2009, 4, e4429. [Google Scholar] [CrossRef] [PubMed]
- Ask, K.; Jasencakova, Z.; Menard, P.; Feng, Y.; Almouzni, G.; Groth, A. Codanin-1, mutated in the anaemic disease CDAI, regulates Asf1 function in S-phase histone supply. EMBO J. 2012, 31, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Reinberg, D.; Sims, R.J. de FACTo nucleosome dynamics. J. Biol. Chem. 2006, 281, 23297–23301. [Google Scholar] [CrossRef]
- Hsieh, F.-K.; Kulaeva, O.I.; Patel, S.S.; Dyer, P.N.; Luger, K.; Reinberg, D.; Studitsky, V.M. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc. Natl. Acad. Sci. USA 2013, 110, 7654–7659. [Google Scholar] [CrossRef]
- Fujiu, K.; Numata, O. Identification and molecular cloning of Tetrahymena 138-kDa protein, a transcription elongation factor homologue that interacts with microtubules in vitro. Biochem. Biophys. Res. Commun. 2004, 315, 196–203. [Google Scholar] [CrossRef]
- Alvarez, F.; Muñoz, F.; Schilcher, P.; Imhof, A.; Almouzni, G.; Loyola, A. Sequential establishment of marks on soluble histones H3 and H4. J. Biol. Chem. 2011, 286, 17714–17721. [Google Scholar] [CrossRef]
- Levy-Wilson, B. Glycosylation, ADP-ribosylation, and methylation of Tetrahymena histones. Biochemistry 1983, 22, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Gjoneska, E.; Ren, Q.; Taverna, S.D.; Allis, C.D.; Gorovsky, M.A. Phosphorylation of the SQ H2A.X motif is required for proper meiosis and mitosis in Tetrahymena thermophila. Mol. Cell. Biol. 2007, 27, 2648–2660. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, B.; Gorovsky, M.A. Histone H2B ubiquitylation is not required for histone H3 methylation at lysine 4 in tetrahymena. J. Biol. Chem. 2009, 284, 34870–34879. [Google Scholar] [CrossRef] [PubMed]
- Stargell, L.A.; Bowen, J.; Dadd, C.A.; Dedon, P.C.; Davis, M.; Cook, R.G.; Allis, C.D.; Gorovsky, M.A. Temporal and spatial association of his tone H2A variant hv1 with transcriptionally competent chromatin during nuclear development in Tetrahymena thermophila. Genes Dev. 1993, 7, 2641–2651. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.D.; Falkowska, K.A.; Li, A.Y.; Galanti, S.E.; Kanuru, R.C.; LaMont, E.G.; Mazzarella, K.C.; Micev, A.J.; Osman, M.M.; Piotrowski, N.K.; et al. Nucleus-specific importin α proteins and nucleoporins regulate protein import and nuclear division in the binucleate Tetrahymena thermophila. Eukaryot. Cell 2008, 7, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.E.; Allis, C.D.; Strahl, B.D. Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 2011, 409, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Biterge, B.; Schneider, R. Histone variants: Key players of chromatin. Cell Tissue Res. 2014, 356, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Chen, G.; Thanos, D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002, 111, 381–392. [Google Scholar] [CrossRef]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef]
- Richters, A.; Koehler, A.N. Epigenetic Modulation Using Small Molecules—Targeting Histone Acetyltransferases in Disease. Curr. Med. Chem. 2017, 24, 4121–4150. [Google Scholar] [CrossRef]
- Baker, S.P.; Grant, P.A. The SAGA continues: Expanding the cellular role of a transcriptional co-activator complex. Oncogene 2007, 26, 5329–5340. [Google Scholar] [CrossRef]
- Allard, S.; Utley, R.T.; Savard, J.; Clarke, A.; Grant, P.; Brandl, C.J.; Pillus, L.; Workman, J.L.; Côté, J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999, 18, 5108–5119. [Google Scholar] [CrossRef] [PubMed]
- Parthun, M.R. Hat1: The emerging cellular roles of a type B histone acetyltransferase. Oncogene 2007, 26, 5319–5328. [Google Scholar] [CrossRef]
- Berndsen, C.E.; Denu, J.M. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 2008, 18, 682–689. [Google Scholar] [CrossRef]
- Jain, A.K.; Barton, M.C. Bromodomain Histone Readers and Cancer. J. Mol. Biol. 2017, 429, 2003–2010. [Google Scholar] [CrossRef]
- Steunou, A.-L.; Cramet, M.; Rossetto, D.; Aristizabal, M.J.; Lacoste, N.; Drouin, S.; Côté, V.; Paquet, E.; Utley, R.T.; Krogan, N.; et al. Combined Action of Histone Reader Modules Regulates NuA4 Local Acetyltransferase Function but Not Its Recruitment on the Genome. Mol. Cell. Biol. 2016, 36, 2768–2781. [Google Scholar] [CrossRef]
- Taverna, S.D.; Ilin, S.; Rogers, R.S.; Tanny, J.C.; Lavender, H.; Li, H.; Baker, L.; Boyle, J.; Blair, L.P.; Chait, B.T.; et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell 2006, 24, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, B. GorovskyMA Essential and nonessential histone H2A variants in Tetrahymena thermophila. Mol. Cell. Biol. 1996, 16, 4305–4311. [Google Scholar] [CrossRef]
- Papamichos-chronakis, M.; Watanabe, S.; Rando, O.J.; Craig, L. Global regulation of H2A.Z localization by the INO80 chromatin remodeling enzyme is essential for genome integrity. Cell 2011, 144, 200–213. [Google Scholar] [CrossRef]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2017, 372, 20160290. [Google Scholar] [CrossRef]
- Vavra, K.J.; Allis, C.D.; Gorovsky, M.A. Regulation of histone acetylation in Tetrahymena macro- and micronuclei. J. Biol. Chem. 1982, 257, 2591–2598. [Google Scholar]
- Hutchcroft, J.E.; Anostario, M.; Harrison, M.L.; Geahlen, R.L. Renaturation and assay of protein kinases after electrophoresis in sodium dodecyl sulfate-polyacrylamide gels. Methods Enzymol. 1991, 200, 417–423. [Google Scholar]
- Guarente, L. Transcriptional coactivators in yeast and beyond. Trends Biochem. Sci. 1995, 20, 517–521. [Google Scholar] [CrossRef]
- Georgakopoulos, T.; Thireos, G. Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription. EMBO J. 1992, 11, 4145–4152. [Google Scholar] [CrossRef]
- Reisman, D.; Glaros, S.; Thompson, E.A. The SWI/SNF complex and cancer. Oncogene 2009, 28, 1653–1668. [Google Scholar] [CrossRef]
- Fillingham, J.; Garg, J.; Tsao, N.; Vythilingum, N.; Nishikawa, T.; Pearlman, R.E. Molecular genetic analysis of an SNF2/brahma-related gene in Tetrahymena thermophila suggests roles in growth and nuclear development. Eukaryot. Cell 2006, 5, 1347–1359. [Google Scholar] [CrossRef]
- Teo, G.; Liu, G.; Zhang, J.; Nesvizhskii, A.I.; Gingras, A.-C.; Choi, H. SAINTexpress: Improvements and additional features in Significance Analysis of INTeractome software. J. Proteom. 2014, 100, 37–43. [Google Scholar] [CrossRef]
- Shi, X.; Hong, T.; Walter, K.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Peña, P.; Lan, F.; Kaadige, M.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef]
- Watanabe, S.; Radman-Livaja, M.; Rando, O.J.; Peterson, C.L. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 2013, 340, 195–199. [Google Scholar] [CrossRef]
- Platt, J.L.; Rogers, B.J.; Rogers, K.C.; Harwood, A.J.; Kimmel, A.R. Different CHD chromatin remodelers are required for expression of distinct gene sets and specific stages during development of Dictyostelium discoideum. Development 2013, 140, 4926–4936. [Google Scholar] [CrossRef]
- Eissenberg, J.C. Structural biology of the chromodomain: Form and function. Gene 2012, 496, 69–78. [Google Scholar] [CrossRef]
- DeRango-Adem, E. Macromolecular Interactome of Tetrahymena CHD Family Chromatin Remodelers. Master’s Thesis, York University, Toronto, ON, Canada, 2017. [Google Scholar]
- Kataoka, K.; Mochizuki, K. Programmed DNA elimination in Tetrahymena: A small RNA-mediated genome surveillance mechanism. Adv. Exp. Med. Biol. 2011, 722, 156–173. [Google Scholar]
- Chalker, D.L.; Yao, M.C. Nongenic, bidirectional transcription precedes and may promote developmental DNA deletion in Tetrahymena thermophila. Genes Dev. 2001, 15, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Gorovsky, M.A. RNA polymerase II localizes in Tetrahymena thermophila meiotic micronuclei when micronuclear transcription associated with genome rearrangement occurs. Eukaryot. Cell 2004, 3, 1233–1240. [Google Scholar] [CrossRef]
- Schoeberl, U.E.; Kurth, H.M.; Noto, T.; Mochizuki, K. Biased transcription and selective degradation of small RNAs shape the pattern of DNA elimination in Tetrahymena. Genes Dev. 2012, 26, 1729–1742. [Google Scholar] [CrossRef] [PubMed]
- Garg, J.; Saettone, A.; Nabeel-Shah, S.; Cadorin, M.; Ponce, M.; Marquez, S.; Pu, S.; Greenblatt, J.; Lambert, J.-P.; Pearlman, R.E.; et al. The MED31 Conserved Component of the Divergent Mediator Complex in Tetrahymena Thermophila Participates in Developmental Regulation. 2019; in submission. [Google Scholar]
- Boekhorst, J.; van Breukelen, B.; Heck, A.; Snel, B. Comparative phosphoproteomics reveals evolutionary and functional conservation of phosphorylation across eukaryotes. Genome Biol. 2008, 9, R144. [Google Scholar] [CrossRef] [PubMed]
- Eirín-López, J.M.; Rebordinos, L.; Rooney, A.P.; Rozas, J. The birth-and-death evolution of multigene families revisited. Genome Dyn. 2012, 7, 170–196. [Google Scholar] [PubMed]
- Frehlick, L.J.; Eirín-López, J.M.; Ausió, J. New insights into the nucleophosmin/nucleoplasmin family of nuclear chaperones. Bioessays 2007, 29, 49–59. [Google Scholar] [CrossRef] [PubMed]
- González-Romero, R.; Eirín-López, J.M.; Ausió, J. Evolution of high mobility group nucleosome-binding proteins and its implications for vertebrate chromatin specialization. Mol. Biol. Evol. 2015, 32, 121–131. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Nabeel-Shah, S.; Ashraf, K.; Pearlman, R.E.; Fillingham, J. Molecular evolution of NASP and conserved histone H3/H4 transport pathway. BMC Evol. Biol. 2014, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ge, Z.; Walsh, S.T.R.; Parthun, M.R. The human histone chaperone sNASP interacts with linker and core histones through distinct mechanisms. Nucleic Acids Res. 2012, 40, 660–669. [Google Scholar] [CrossRef]
- Dannah, N.S.; Nabeel-Shah, S.; Kurat, C.F.; Sabatinos, S.A.; Fillingham, J. Functional Analysis of Hif1 Histone Chaperone in Saccharomyces cerevisiae. G3 (Bethesda) 2018, 8, 1993–2006. [Google Scholar] [CrossRef]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef]
- Tian, M.; Chen, X.; Xiong, Q.; Xiong, J.; Xiao, C.; Ge, F.; Yang, F.; Miao, W. Phosphoproteomic Analysis of Protein Phosphorylation Networks in Tetrahymena thermophila, a Model Single-celled Organism. Mol. Cell. Proteom. 2014, 13, 503–519. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef]
- Gupta, G.D.; Coyaud, É.; Gonçalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.T.; et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Uusküla-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II β interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [PubMed]
- Youn, J.-Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-P.; Picaud, S.; Fujisawa, T.; Hou, H.; Savitsky, P.; Uusküla-Reimand, L.; Gupta, G.D.; Abdouni, H.; Lin, Z.-Y.; Tucholska, M.; et al. Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol. Cell 2019, 73, 621–638.e17. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-P.; Tucholska, M.; Go, C.; Knight, J.D.R.; Gingras, A.-C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteom. 2015, 118, 81–94. [Google Scholar] [CrossRef]
- Louka, P.; Vasudevan, K.K.; Guha, M.; Joachimiak, E.; Wloga, D.; Tomasi, R.F.-X.; Baroud, C.N.; Dupuis-Williams, P.; Galati, D.F.; Pearson, C.G.; et al. Proteins that control the geometry of microtubules at the ends of cilia. J. Cell Biol. 2018, 217, 4298–4313. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, P.; Joachimiak, E.; Bazan, R.; Fu, G.; Poprzeczko, M.; Fabczak, H.; Nicastro, D.; Wloga, D. Ciliary proteins Fap43 and Fap44 interact with each other and are essential for proper cilia and flagella beating. Cell. Mol. Life Sci. 2018, 75, 4479–4493. [Google Scholar] [CrossRef] [PubMed]
- Janetopoulos, C.; Cole, E.; Smothers, J.F.; Allis, C.D.; Aufderheide, K.J. The conjusome: A novel structure in Tetrahymena found only during sexual reorganization. J. Cell Sci. 1999, 112 Pt 7, 1003–1011. [Google Scholar]
- Xu, J.; Yuan, Y.; Liang, A.; Wang, W. Chromodomain protein Tcd1 is required for macronuclear genome rearrangement and repair in Tetrahymena. Sci. Rep. 2015, 5, 10243. [Google Scholar] [CrossRef]
- Decker, C.J.; Parker, R. P-bodies and stress granules: Possible roles in the control of translation and mRNA degradation. Cold Spring Harb. Perspect. Biol. 2012, 4, a012286. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, K.M. Special focus on the Cajal Body. RNA Biol. 2017, 14, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Wright, J.; Peckner, R.; Kalish, B.T.; Zhang, F.; Carr, S.A. Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat. Methods 2018, 15, 437–439. [Google Scholar] [CrossRef]
- Gao, X.D.; Tu, L.-C.; Mir, A.; Rodriguez, T.; Ding, Y.; Leszyk, J.; Dekker, J.; Shaffer, S.A.; Zhu, L.J.; Wolfe, S.A.; et al. C-BERST: Defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat. Methods 2018, 15, 433–436. [Google Scholar] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saettone, A.; Nabeel-Shah, S.; Garg, J.; Lambert, J.-P.; Pearlman, R.E.; Fillingham, J. Functional Proteomics of Nuclear Proteins in Tetrahymena thermophila: A Review. Genes 2019, 10, 333. https://doi.org/10.3390/genes10050333
Saettone A, Nabeel-Shah S, Garg J, Lambert J-P, Pearlman RE, Fillingham J. Functional Proteomics of Nuclear Proteins in Tetrahymena thermophila: A Review. Genes. 2019; 10(5):333. https://doi.org/10.3390/genes10050333
Chicago/Turabian StyleSaettone, Alejandro, Syed Nabeel-Shah, Jyoti Garg, Jean-Philippe Lambert, Ronald E. Pearlman, and Jeffrey Fillingham. 2019. "Functional Proteomics of Nuclear Proteins in Tetrahymena thermophila: A Review" Genes 10, no. 5: 333. https://doi.org/10.3390/genes10050333
APA StyleSaettone, A., Nabeel-Shah, S., Garg, J., Lambert, J.-P., Pearlman, R. E., & Fillingham, J. (2019). Functional Proteomics of Nuclear Proteins in Tetrahymena thermophila: A Review. Genes, 10(5), 333. https://doi.org/10.3390/genes10050333