Genome-Wide Investigation and Functional Analysis of Sus scrofa RNA Editing Sites across Eleven Tissues



Abstract
1. Introduction
2. Materials and Methods
2.1. Reads Mapping and RNA-Editing Sites Identification
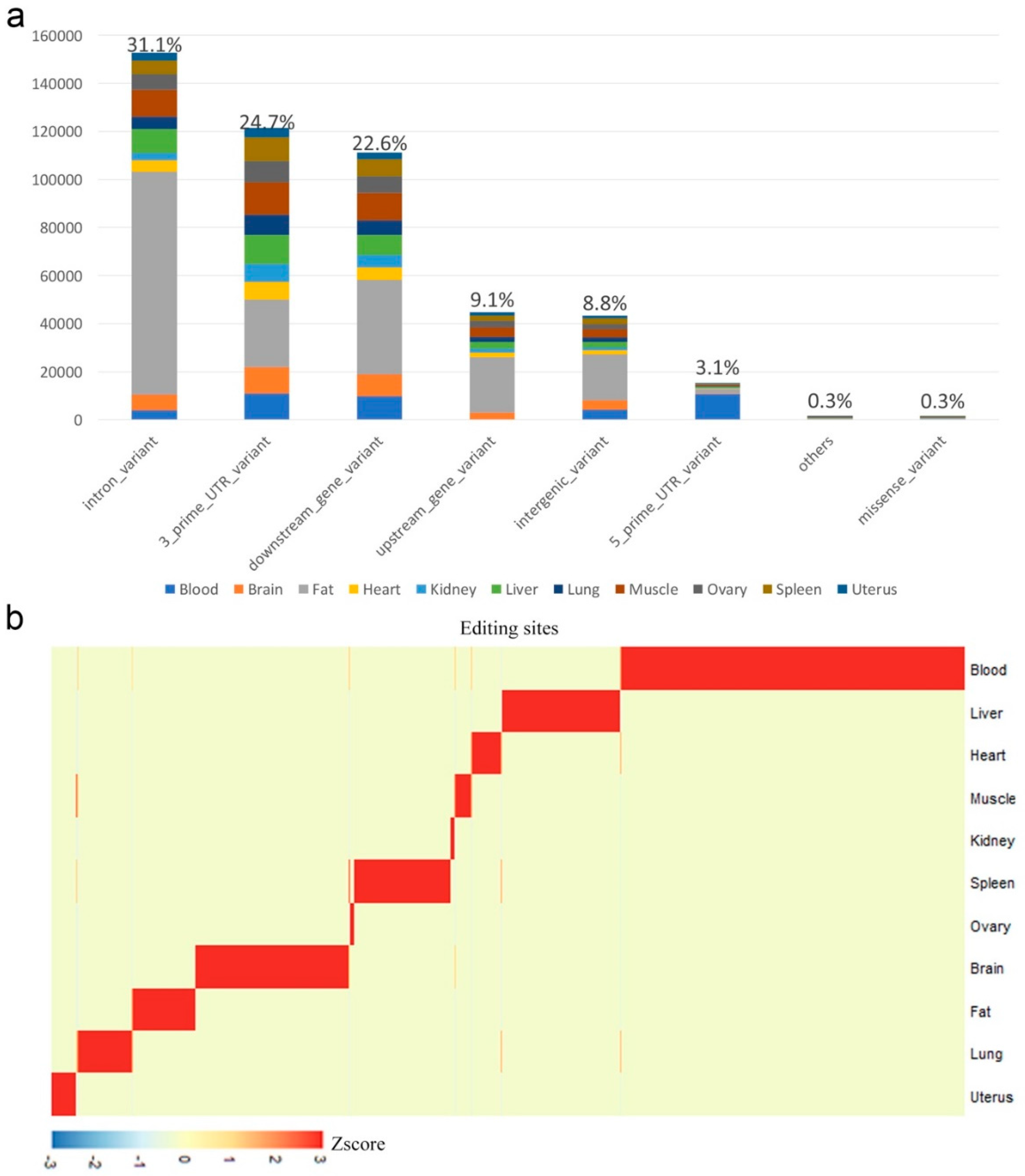
2.2. Tissue-Specific Editing
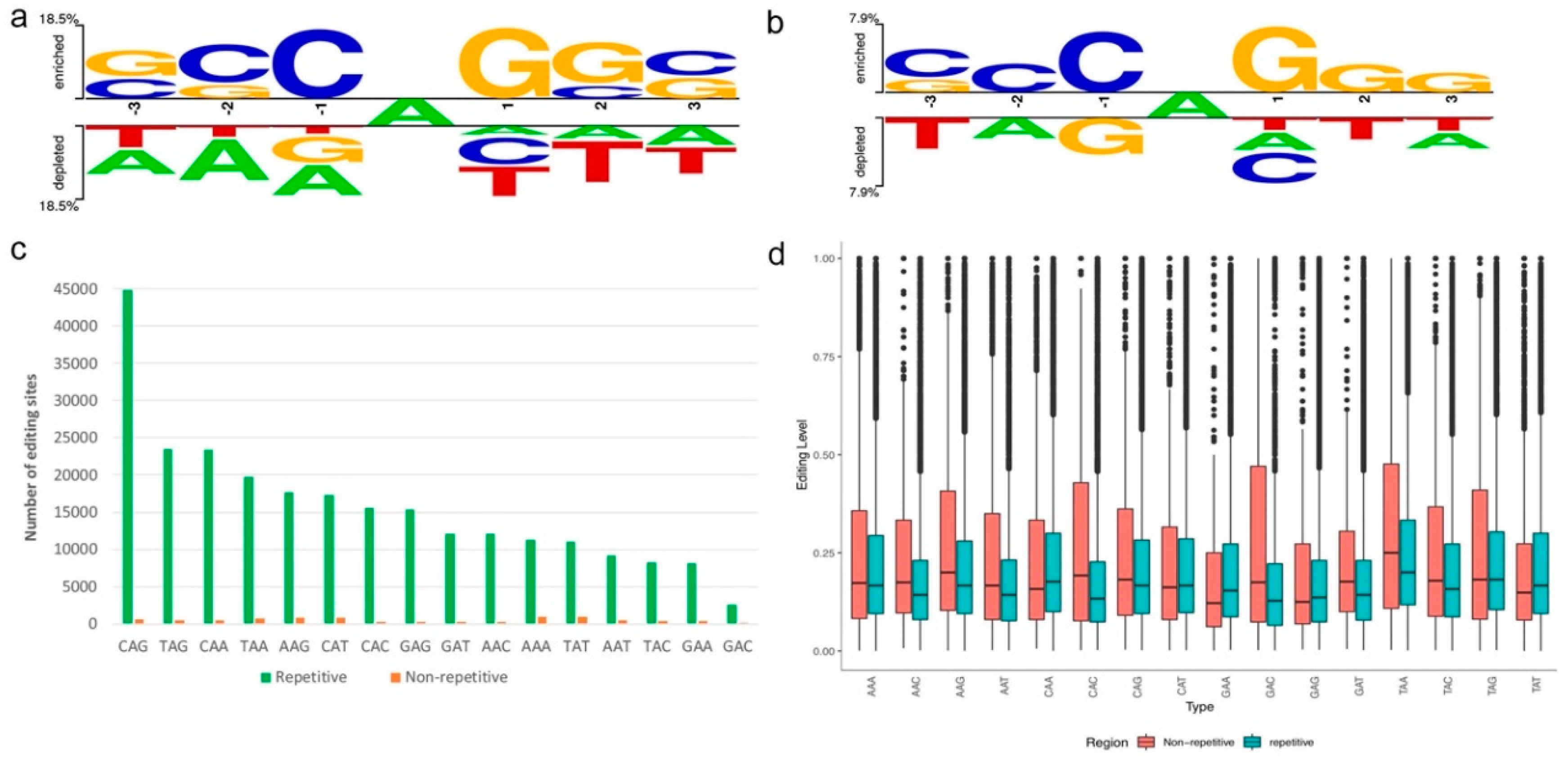
2.3. Sequence Preference
2.4. Genomic Feature Annotation
2.5. Impacts of RNA Editing Sites on miRNA–mRNA Interactions
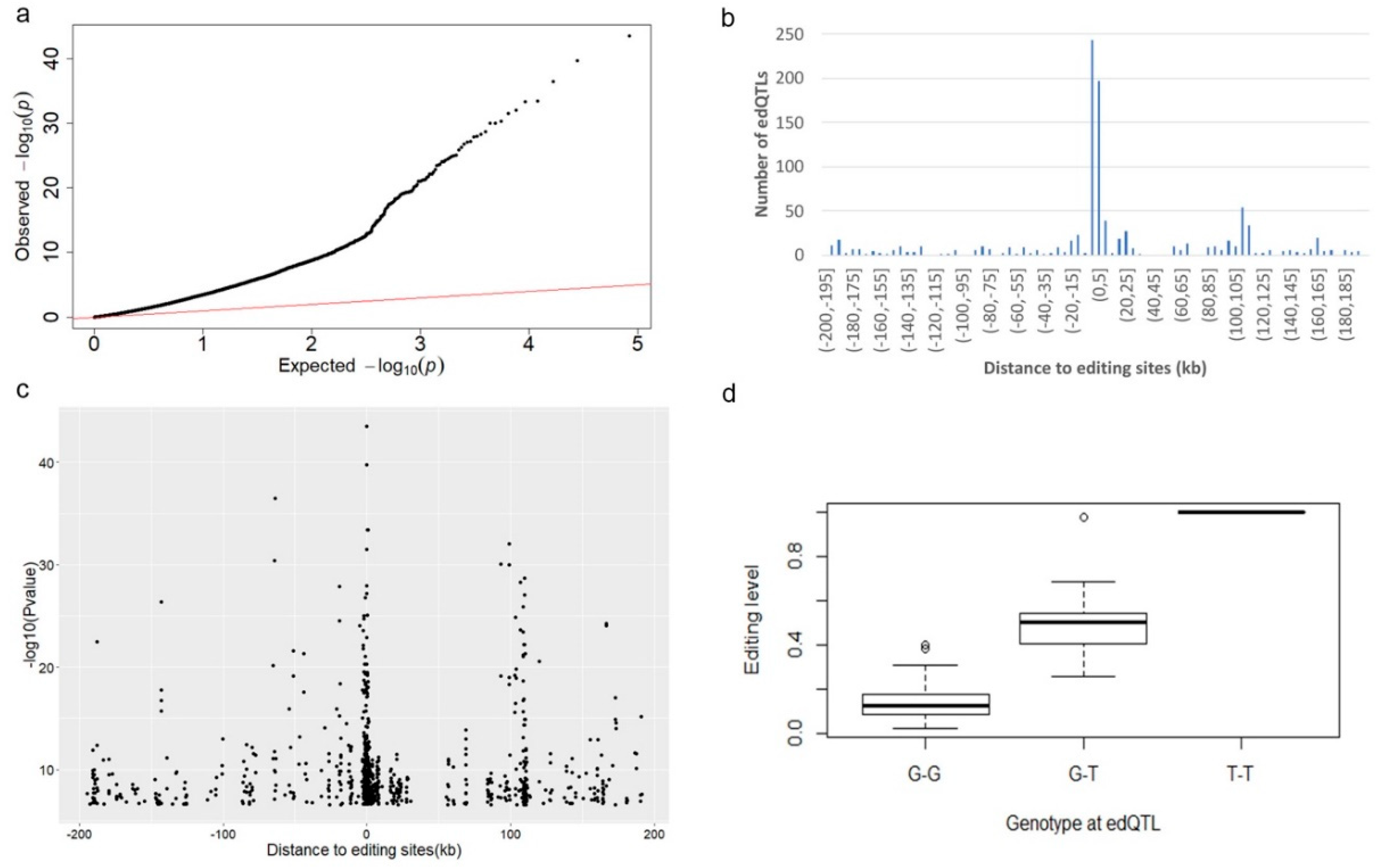
2.6. edQTL Analysis
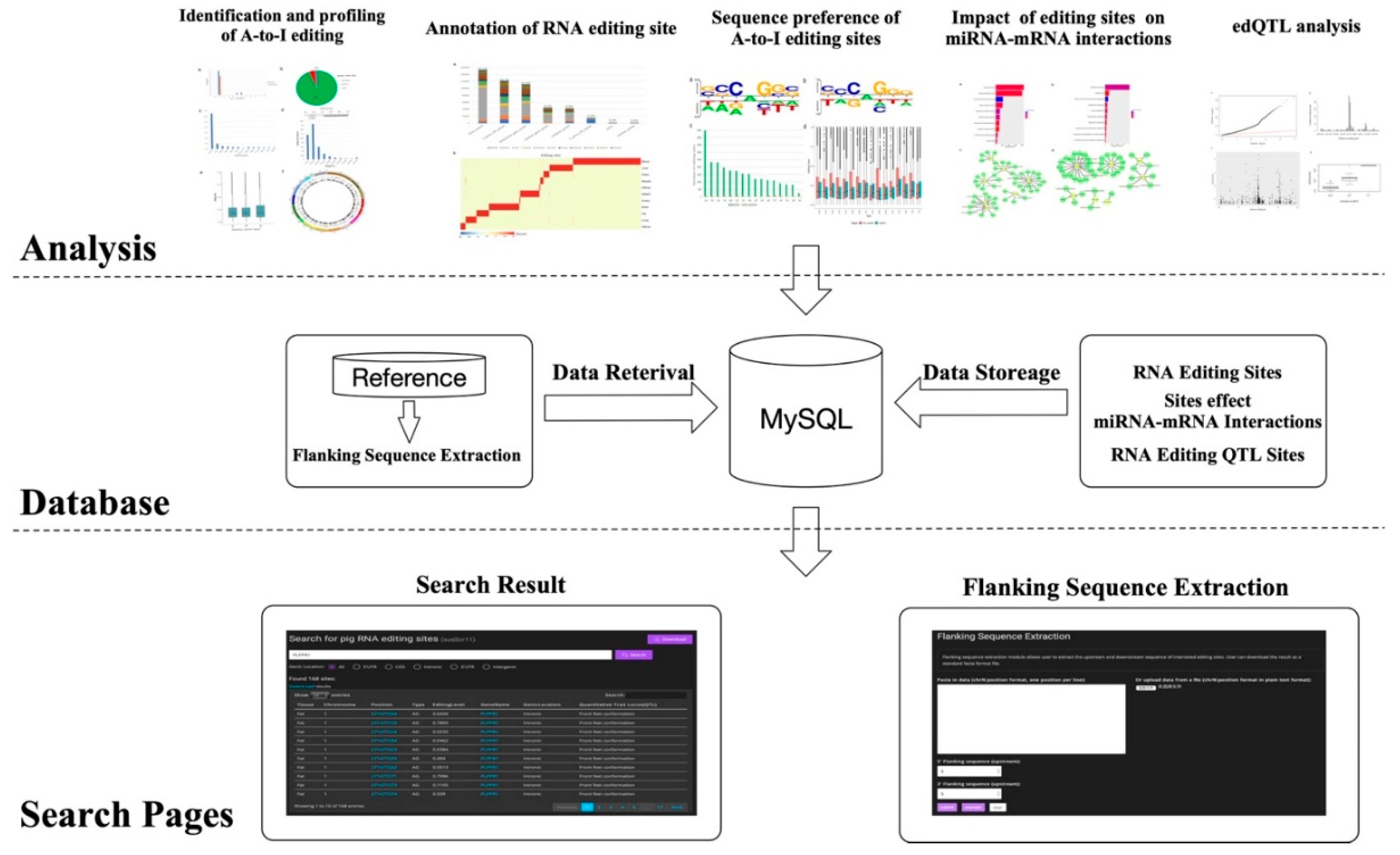
2.7. Database Construction
3. Results
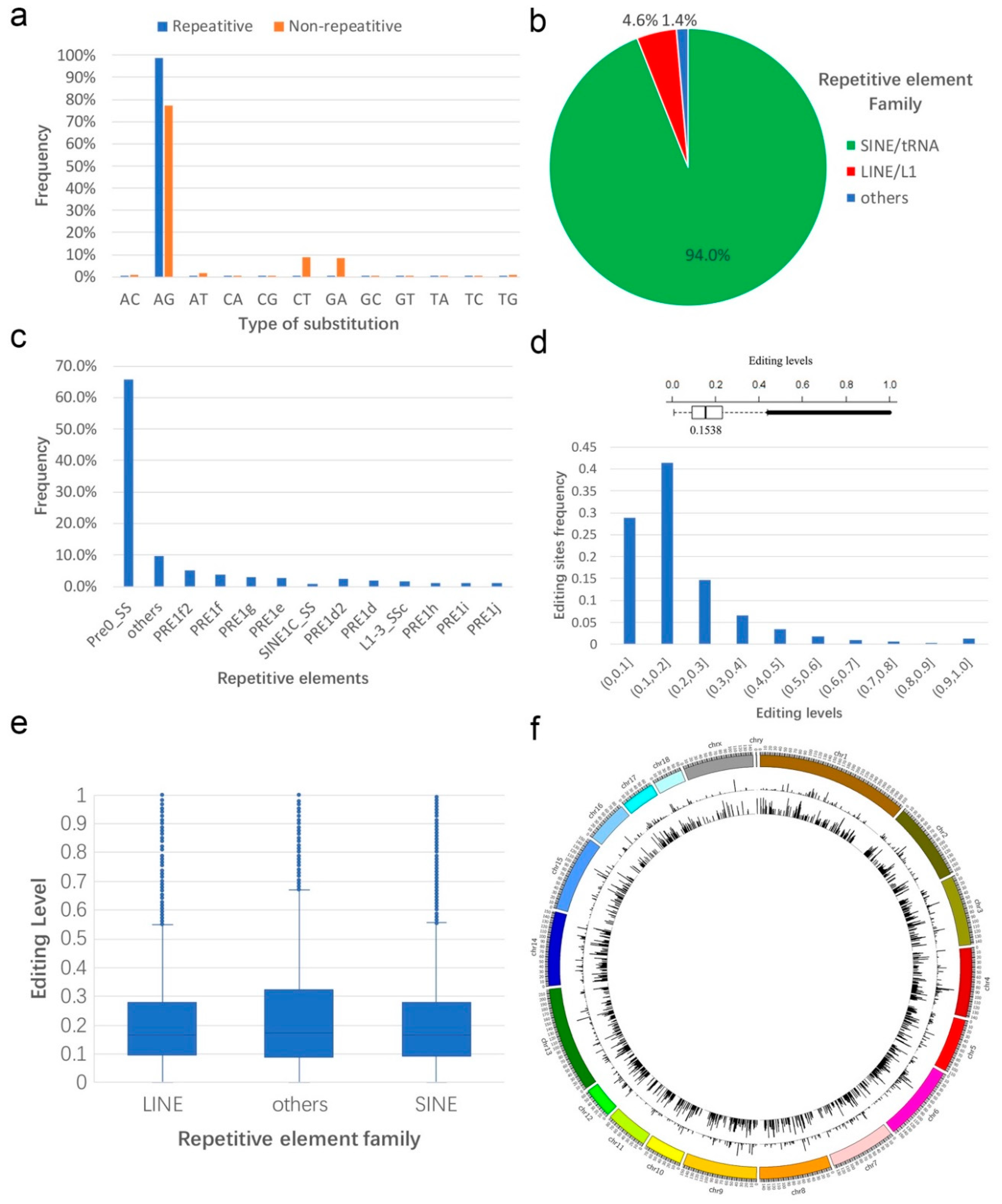
3.1. Identification and Profiling of Sus scrofa RNA Editing Sites
3.2. Annotation of RNA Editing Sites
3.3. Sequence Preference of A-to-I Editing Sites
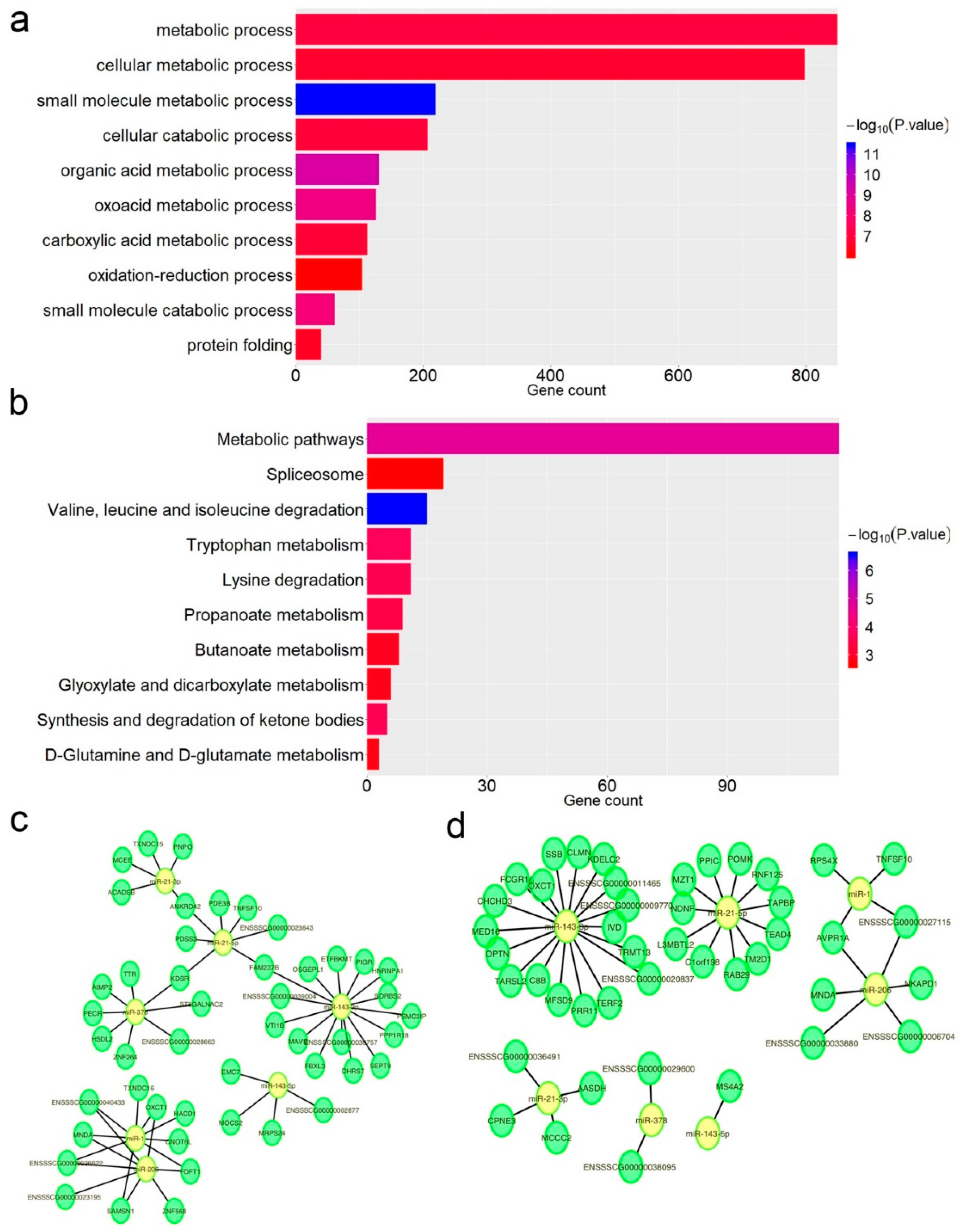
3.4. Impact of A-to-I Editing on miRNA–mRNA Interactions
3.5. edQTL Analysis
3.6. Sus scrofa RNA Editing Sites Database
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef]
- Nishikura, K. Functions and regulation of RNA editing by adar deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Gott, J.M.; Emeson, R.B. Functions and mechanisms of RNA editing. Annu. Rev. Genet. 2000, 34, 499–531. [Google Scholar] [CrossRef]
- Lee, J.-H.; Ang, J.K.; Xiao, X. Analysis and design of RNA sequencing experiments for identifying RNA editing and other single-nucleotide variants. RNA 2013, 19, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Miyakoda, M.; Yang, W.; Khillan, J.; Stachura, D.L.; Weiss, M.J.; Nishikura, K. Stress-induced apoptosis associated with null mutation of adar1 RNA editing deaminase gene. J. Biol. Chem. 2004, 279, 4952–4961. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an ampa receptor gene rescues lethality in mice deficient in the RNA-editing enzyme adar2. Nature 2000, 406, 78. [Google Scholar] [CrossRef]
- Sommer, B.; Köhler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Köhler, M.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: Diversity by RNA editing. Neuron 1993, 10, 491–500. [Google Scholar] [CrossRef]
- Lomeli, H.; Mosbacher, J.; Melcher, T.; Hoger, T.; Kuner, T.; Monyer, H.; Higuchi, M.; Bach, A.; Seeburg, P.H. Control of kinetic properties of ampa receptor channels by nuclear RNA editing. Science 1994, 266, 1709–1713. [Google Scholar] [CrossRef]
- Burns, C.M.; Chu, H.; Rueter, S.M.; Hutchinson, L.K.; Canton, H.; Sanders-Bush, E.; Emeson, R.B. Regulation of serotonin-2c receptor g-protein coupling by RNA editing. Nature 1997, 387, 303. [Google Scholar] [CrossRef]
- Casey, J.L.; Gerin, J.L. Hepatitis d virus RNA editing: Specific modification of adenosine in the antigenomic RNA. J. Virol. 1995, 69, 7593–7600. [Google Scholar]
- Poison, A.G.; Bass, B.L.; Casey, J.L. RNA editing of hepatitis delta virus antigenome by dsrna-adenosine deaminase. Nature 1996, 380, 454. [Google Scholar] [CrossRef]
- Blow, M.J.; Grocock, R.J.; van Dongen, S.; Enright, A.J.; Dicks, E.; Futreal, P.A.; Wooster, R.; Stratton, M.R. RNA editing of human micrornas. Genome Biol. 2006, 7, R27. [Google Scholar] [CrossRef]
- Kume, H.; Hino, K.; Galipon, J.; Ui-Tei, K. A-to-i editing in the mirna seed region regulates target mrna selection and silencing efficiency. Nucleic Acids Res. 2014, 42, 10050–10060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, C.-S.; Varelas, X.; Monti, S. Altered RNA editing in 3′ utr perturbs microrna-mediated regulation of oncogenes and tumor-suppressors. Sci. Rep. 2016, 6, 23226. [Google Scholar] [CrossRef]
- Prasanth, K.V.; Prasanth, S.G.; Xuan, Z.; Hearn, S.; Freier, S.M.; Bennett, C.F.; Zhang, M.Q.; Spector, D.L. Regulating gene expression through RNA nuclear retention. Cell 2005, 123, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A. Mutations in adar1 cause aicardi-goutieres syndrome associated with a type i interferon signature. Nat. Genet. 2012, 44, 1243. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Kwak, S. The molecular link between inefficient glua2 q/r site-RNA editing and tdp-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res. 2014, 1584, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Eran, A.; Li, J.B.; Vatalaro, K.; McCarthy, J.; Rahimov, F.; Collins, C.; Markianos, K.; Margulies, D.M.; Brown, E.N.; Calvo, S.E. Comparative RNA editing in autistic and neurotypical cerebella. Mol. Psychiatry 2013, 18, 1041. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toth, A.M.; Li, Z.; Cattaneo, R.; Samuel, C.E. RNA-specific adenosine deaminase adar1 suppresses measles virus-induced apoptosis and activation of protein kinase pkr. J. Biol. Chem. 2009, 284, 29350–29356. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.; Eterovic, A.K.; Yuan, Y. The genomic landscape and clinical relevance of a-to-i RNA editing in human cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef]
- Martínez-Montes, A.; Fernández, A.; Pérez-Montarelo, D.; Alves, E.; Benítez, R.; Nuñez, Y.; Ovilo, C.; Ibañez-Escriche, N.; Folch, J.; Fernandez, A. Using RNA-seq snp data to reveal potential causal mutations related to pig production traits and RNA editing. Anim. Genet. 2017, 48, 151–165. [Google Scholar] [CrossRef]
- Funkhouser, S.A.; Steibel, J.P.; Bates, R.O.; Raney, N.E.; Schenk, D.; Ernst, C.W. Evidence for transcriptome-wide RNA editing among sus scrofa pre-1 sine elements. Bmc Genom. 2017, 18, 360. [Google Scholar] [CrossRef]
- Stulić, M.; Jantsch, M.F. Spatio-temporal profiling of filamin a RNA-editing reveals adar preferences and high editing levels outside neuronal tissues. RNA Biol. 2013, 10, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Neeman, Y.; Levanon, E.Y.; Jantsch, M.F.; Eisenberg, E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 2006, 12, 1802–1809. [Google Scholar] [CrossRef]
- Picardi, E.; Regina, T.M.R.; Brennicke, A.; Quagliariello, C. Redidb: The RNA editing database. Nucleic Acids Res. 2006, 35, D173–D177. [Google Scholar] [CrossRef][Green Version]
- Kiran, A.; Baranov, P.V. Darned: A database of RNA editing in humans. Bioinformatics 2010, 26, 1772–1776. [Google Scholar] [CrossRef]
- Ramaswami, G.; Li, J.B. Radar: A rigorously annotated database of a-to-i RNA editing. Nucleic Acids Res. 2013, 42, D109–D113. [Google Scholar] [CrossRef]
- Picardi, E.; D’Erchia, A.M.; Lo Giudice, C.; Pesole, G. Rediportal: A comprehensive database of a-to-i RNA editing events in humans. Nucleic Acids Res. 2016, 45, D750–D757. [Google Scholar] [CrossRef]
- Kumar, S.; Hedges, S.B. A molecular timescale for vertebrate evolution. Nature 1998, 392, 917. [Google Scholar] [CrossRef]
- Larson, G.; Dobney, K.; Albarella, U.; Fang, M.; Matisoo-Smith, E.; Robins, J.; Lowden, S.; Finlayson, H.; Brand, T.; Willerslev, E. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 2005, 307, 1618–1621. [Google Scholar] [CrossRef]
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393. [Google Scholar] [CrossRef]
- Wernersson, R.; Schierup, M.H.; Jørgensen, F.G.; Gorodkin, J.; Panitz, F.; Stærfeldt, H.-H.; Christensen, O.F.; Mailund, T.; Hornshøj, H.; Klein, A. Pigs in sequence space: A 0.66 x coverage pig genome survey based on shotgun sequencing. BMC Genom. 2005, 6, 70. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Y.; Yang, J.-Y.; Cheng, D.; Liu, X.; Ma, X.; West, F.D.; Wang, H. Comparative gene expression signature of pig, human and mouse induced pluripotent stem cell lines reveals insight into pig pluripotency gene networks. Stem Cell Rev. Rep. 2014, 10, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Wall, R.; Shani, M. Are animal models as good as we think? Theriogenology 2008, 69, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P. Ensembl 2017. Nucleic Acids Res. 2016, 45, D635–D642. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for snp calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Zhang, F.; Lu, Y.; Yan, S.; Xing, Q.; Tian, W. Sprint: An snp-free toolkit for identifying rna editing sites. Bioinformatics (Oxford, England) 2017, 33, 3538–3548. [Google Scholar]
- Kadota, K.; Ye, J.; Nakai, Y.; Terada, T.; Shimizu, K. Roku: A novel method for identification of tissue-specific genes. Bmc Bioinform. 2006, 7, 294. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two sample logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Developmental progress and current status of the animal qtldb. Nucleic Acids Res. 2015, 44, D827–D833. [Google Scholar] [CrossRef]
- Krüger, J.; Rehmsmeier, M. Rnahybrid: Microrna target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microrna. Org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Ramaswami, G.; Lin, W.; Piskol, R.; Tan, M.H.; Davis, C.; Li, J.B. Accurate identification of human alu and non-alu RNA editing sites. Nat. Methods 2012, 9, 579. [Google Scholar] [CrossRef]
- Li, J.B.; Levanon, E.Y.; Yoon, J.-K.; Aach, J.; Xie, B.; LeProust, E.; Zhang, K.; Gao, Y.; Church, G.M. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science (New York N. Y.) 2009, 324, 1210–1213. [Google Scholar] [CrossRef]
- Ramaswami, G.; Zhang, R.; Piskol, R.; Keegan, L.P.; Deng, P.; O’connell, M.A.; Li, J.B. Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 2013, 10, 128. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff: Snps in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar]
- Pinto, Y.; Cohen, H.Y.; Levanon, E.Y. Mammalian conserved adar targets comprise only a small fragment of the human editosome. Genome Biol. 2014, 15, R5. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microrna-1 and microrna-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228. [Google Scholar]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T. Obesity-induced overexpression of mirna-143 inhibits insulin-stimulated akt activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J. Microrna-206: The skeletal muscle-specific myomir. Biochim. Et Biophys. Acta Gene Regul. Mech. 2008, 1779, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Liang, R.; Yang, Y.; Hou, X.; Wang, Z.; Zhu, S.; Wang, C.; Tang, Z.; Li, K. Microrna-21 regulates pi3k/akt/mtor signaling by targeting tgfβi during skeletal muscle development in pigs. PLoS ONE 2015, 10, e0119396. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Bommer, G.T.; McCoin, C.S.; Sousa, K.M.; Krishnan, V.; MacDougald, O.A. Roles for mirna-378/378* in adipocyte gene expression and lipogenesis. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E198–E206. [Google Scholar] [CrossRef]
- Montgomery, S.B.; Sammeth, M.; Gutierrez-Arcelus, M.; Lach, R.P.; Ingle, C.; Nisbett, J.; Guigo, R.; Dermitzakis, E.T. Transcriptome genetics using second generation sequencing in a caucasian population. Nature 2010, 464, 773. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; AC‘t Hoen, P.; Monlong, J.; Rivas, M.A.; Gonzalez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantitative Trait Locus (QTL) | Number | Percentage |
|---|---|---|
| Conformation score | 7536 | 53.01% |
| Front feet conformation | 2431 | 17.10% |
| Maternal infanticide | 795 | 5.59% |
| Ear weight | 604 | 4.25% |
| Rhinitis | 569 | 4.00% |
| Hind leg conformation | 475 | 3.34% |
| Vertebra number | 442 | 3.11% |
| Spinal curvature | 422 | 2.97% |
| Left teat number | 414 | 2.91% |
| Lumbar vertebra number | 257 | 1.81% |
| Time spent feeding | 188 | 1.32% |
| Ear erectness | 54 | 0.38% |
| Ear size | 15 | 0.11% |
| Umbilical hernia | 11 | 0.08% |
| Thoracic vertebra number | 2 | 0.01% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Feng, X.; Tang, Z.; Li, S.C. Genome-Wide Investigation and Functional Analysis of Sus scrofa RNA Editing Sites across Eleven Tissues. Genes 2019, 10, 327. https://doi.org/10.3390/genes10050327
Wang Z, Feng X, Tang Z, Li SC. Genome-Wide Investigation and Functional Analysis of Sus scrofa RNA Editing Sites across Eleven Tissues. Genes. 2019; 10(5):327. https://doi.org/10.3390/genes10050327
Chicago/Turabian StyleWang, Zishuai, Xikang Feng, Zhonglin Tang, and Shuai Cheng Li. 2019. "Genome-Wide Investigation and Functional Analysis of Sus scrofa RNA Editing Sites across Eleven Tissues" Genes 10, no. 5: 327. https://doi.org/10.3390/genes10050327
APA StyleWang, Z., Feng, X., Tang, Z., & Li, S. C. (2019). Genome-Wide Investigation and Functional Analysis of Sus scrofa RNA Editing Sites across Eleven Tissues. Genes, 10(5), 327. https://doi.org/10.3390/genes10050327