Going Deeper into High and Low Phylogenetic Relationships of Protura

 ,
, 
 , ,
, , 

Abstract

1. Introduction
2. Materials and Methods
2.1. Genome Amplification, Sequencing and Annotation
2.2. Nucleotide Composition
2.3. Mitogenomic Phylogeny
2.4. Phylogenetic Multi-Locus Analyses
2.5. Discovery Methods of Species Delimitation
2.6. Validation Method of Species Delimitation
3. Results
3.1. Molecular Features of Proturan mtDNAs
3.2. Nucleotide Composition of mtDNA Strands
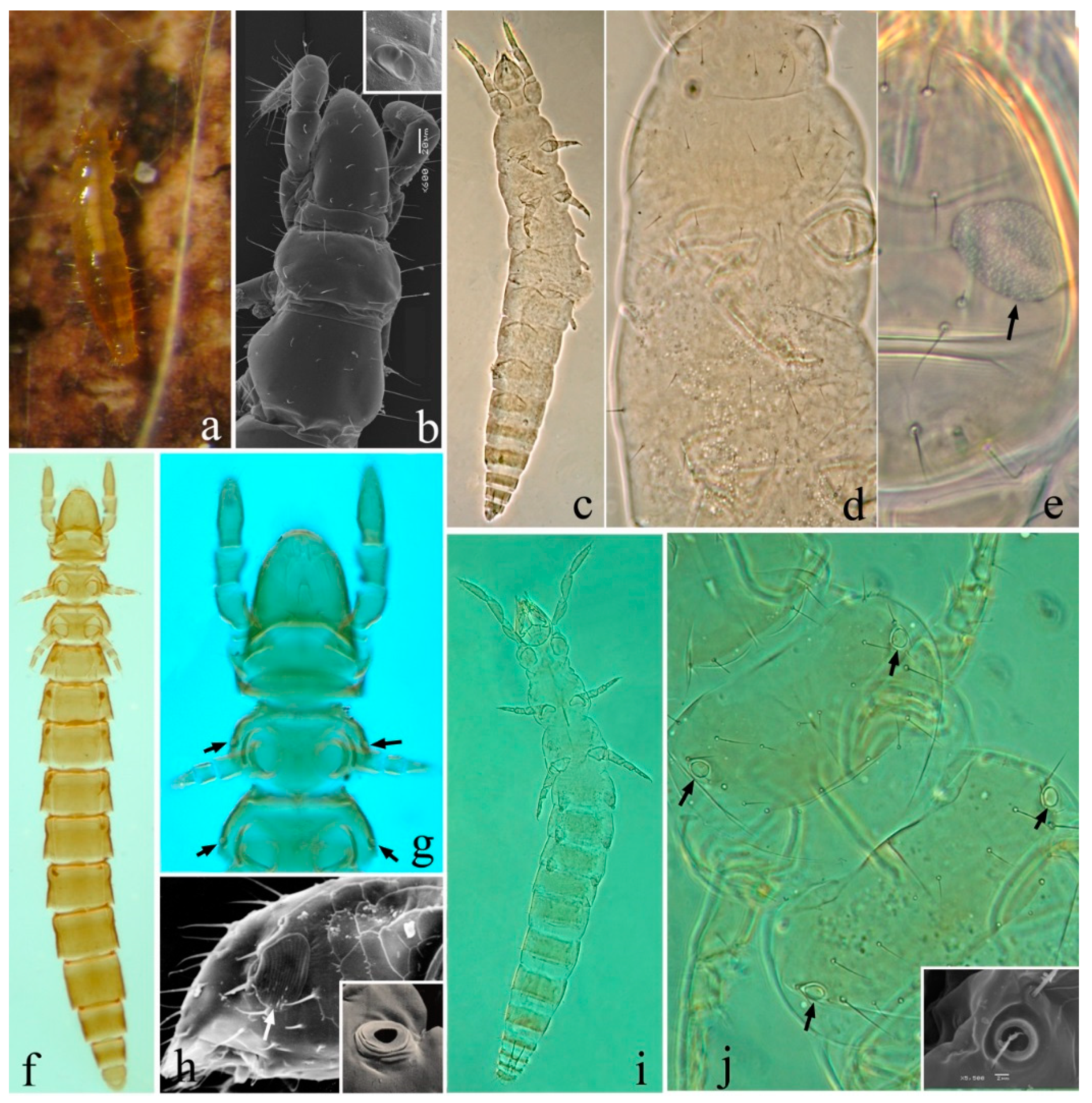
3.3. TRNA Encoding Genes
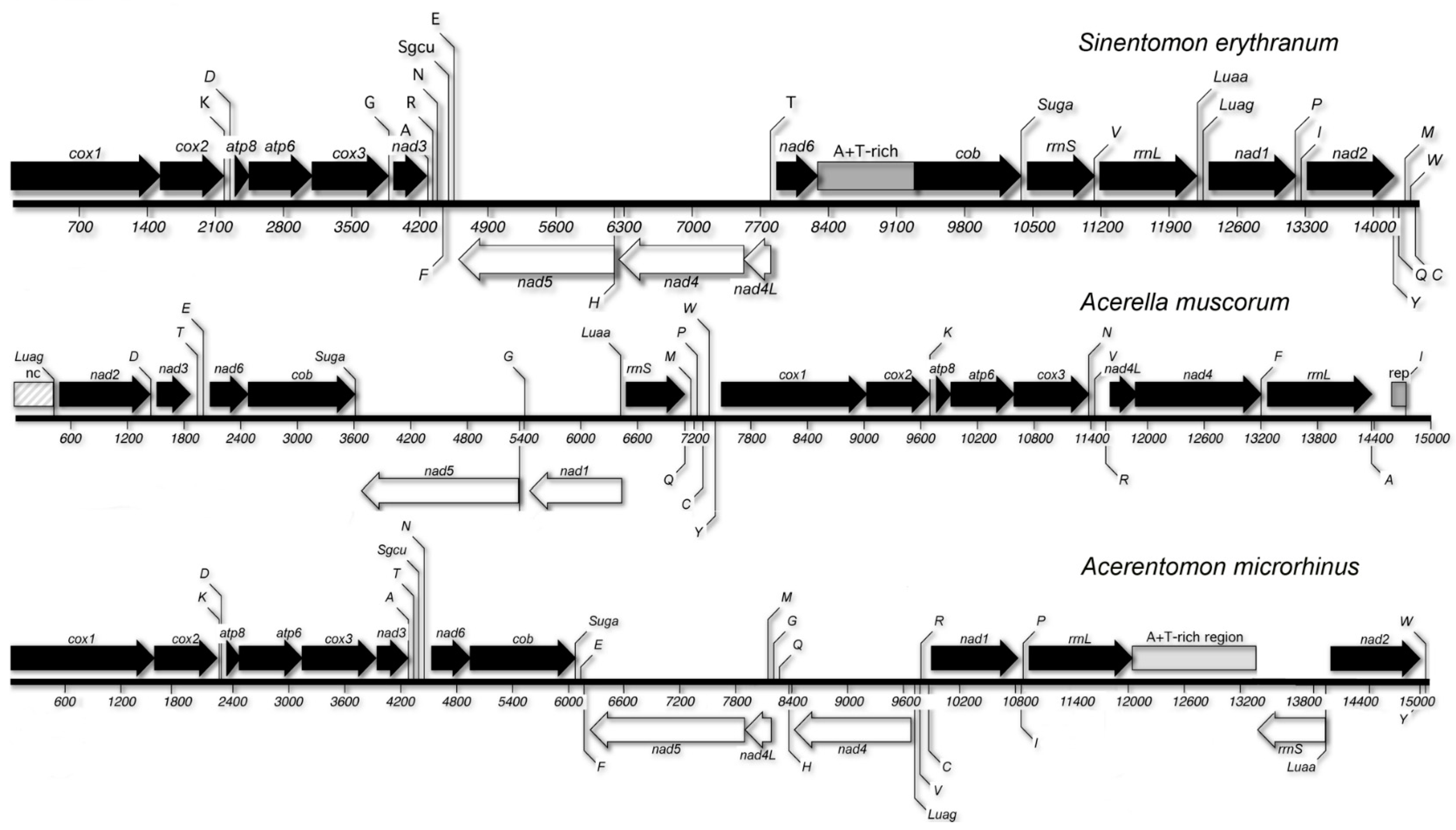
3.4. Gene Order
3.5. Phylogenetic Analyses of Protura in the Context of Arthropoda
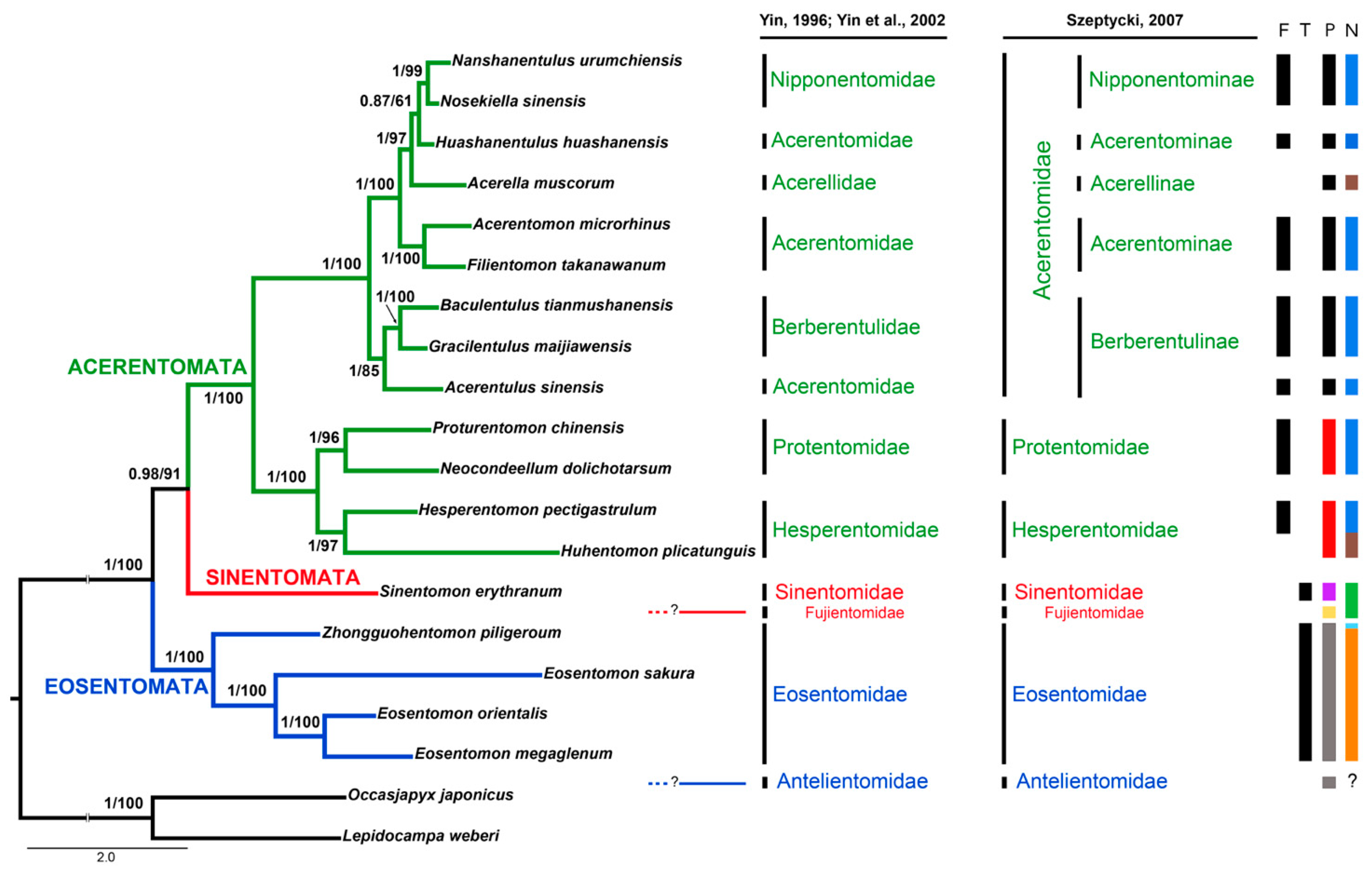
3.6. Phylogenetic Analyses of Protura
3.7. Species Delimitation Analysis
4. Discussion
4.1. Gene Order and Class Relationships
4.2. Compositional Nucleotide Bias
4.3. Aberrant tRNA Genes
4.4. Mitogenomic Phylogenetics
4.5. Internal Relationships at Inter-Order/Family Levels
4.6. Overview on Current Proturan Species Delimitation Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carapelli, A.; Nardi, F.; Dallai, R.; Frati, F. A review of molecular data for the phylogeny of basal hexapods. Pedobiologia 2006, 50, 191–204. [Google Scholar] [CrossRef]
- Szeptycki, A. Catalogue of the World Protura. Acta Zool. Crac. 2007, 50, 1–210. [Google Scholar]
- Bu, Y.; Gao, Y.; Luan, Y.X.; Yin, W.Y. Progress on the systematic study of basal Hexapoda. Chin. Bull. Life Sci. 2012, 24, 130–138. [Google Scholar]
- Bu, Y.; Qian, C.Y.; Luan, Y.X. Three newly recorded species of Acerentomata (Hexapoda: Protura) from China, with analysis of DNA barcodes. Entomotaxonomia 2017, 39, 1–14. [Google Scholar]
- Galli, L.; Shrubovych, J.; Bu, Y.; Zinni, M. Genera of the Protura of the World: Diagnosis, distribution, and key. ZooKeys 2018, 772, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, F. Descrizione di un novo genere d’insetti apterigoti rappresentante di un novo ordine; Bollettino del Laboratorio di Zoologia generale e agraria della R. Scuola superiore d’Agricoltura: Portici, Italia, 1907; pp. 296–311. [Google Scholar]
- Berlese, A. Monografia dei Myrientomata. Redia 1909, 6, 1–182. [Google Scholar]
- Hennig, W. Insect Phylogeny. Translated and Edited by Adrian C. Pont, Revisionary Notes by Dieter Schlee and 9 Collaborators; John Wiley and Sons: New York, NY, USA, 1981; pp. 1–514. [Google Scholar]
- Yin, W.Y. A new idea on phylogeny of Protura with approach to its origin and systematic position. Sci. Sin. Ser. B 1984, 27, 149–160. [Google Scholar]
- Börner, C. Die phylogenetische Bedeutung der Protura. Biologisches Centralblatt 1910, 30, 633–641. [Google Scholar]
- Tuxen, S.L. The Protura. A Revision of the Species of the World. With Keys for Determination; Hermann: Paris, France, 1964; p. 360. [Google Scholar]
- Kristensen, N.P. Phylogeny of insect orders. Annu. Rev. Entomol. 1981, 26, 135–157. [Google Scholar] [CrossRef]
- Dallai, R. Are Protura really Insects? In The Early Evolution of Metazoa and the Significance of Problematic Taxa; Simonetta, A.M., Morris, S.C., Eds.; Cambridge University Press: Cambridge, UK, 1991; pp. 263–269. [Google Scholar]
- Yin, W.Y.; Xue, L.Z. Comparative spermatology of Protura and its significance on proturan systematics. Sci. China Ser. B 1993, 36, 575–586. [Google Scholar]
- Bitsch, C.; Bitsch, J. The phylogenetic interrelationships of the higher taxa of apterygote hexapods. Zool. Scr. 2000, 29, 131–156. [Google Scholar] [CrossRef]
- Yin, W.Y.; Xie, R.D.; Luan, Y.X. On the validity of the class Ellipura (= Parainsecta) (Hexapoda) based on the phylogenetic relationship between Collembola and Protura. Acta Entomol. Sin. 2004, 47, 821–829. [Google Scholar]
- Dallai, R.; Mercati, D.; Bu, Y.; Yin, Y.W. Spermatogenesis and sperm structure of Acerella muscorum, (Ionescu, 1930) (Hexapoda, Protura). Tissue Cell 2010, 42, 97–104. [Google Scholar] [CrossRef]
- Luan, Y.X.; Mallatt, J.M.; Xie, R.D.; Yang, Y.M.; Yin, W.Y. The Phylogenetic Positions of Three Basal-Hexapod Groups (Protura, Diplura, and Collembola) Based on Ribosomal RNA Gene Sequences. Mol. Biol. Evol. 2005, 22, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Mallatt, J.M.; Giribet, G. Further use of nearly complete 28S and 18S rRNA genes to classify Ecdysozoa: 37 more arthropods and a kinorhynch. Mol. Phylogenet. Evol. 2006, 40, 772–794. [Google Scholar] [CrossRef] [PubMed]
- Mallatt, J.M.; Craig, C.W.; Yoder, M.J. Nearly complete rRNA genes assembled from across the metazoan animals: Effects of more taxa, a structure-based alignment, and paired-sites evolutionary models on phylogeny reconstruction. Mol. Phylogenet. Evol. 2010, 55, 1–17. [Google Scholar] [CrossRef]
- Meusemann, K.; von Reumont, B.M.; Simon, S.; Roeding, F.; Strauss, S.; Kück, P.; Ebersberger, I.; Walzl, M.; Pass, G.; Breuers, S.; et al. A Phylogenomic Approach to Resolve the Arthropod Tree of Life. Mol. Biol. Evol. 2010, 27, 2451–2464. [Google Scholar] [CrossRef] [PubMed]
- Machida, R. Evidence from embryology for reconstructing the relationships of hexapod basal clades. Arthropod Syst. Phylogeny 2006, 64, 95–104. [Google Scholar]
- Chen, W.J.; Bu, Y.; Carapelli, A.; Dallai, R.; Li, S.; Yin, W.Y.; Luan, Y.X. Mitochondrial genome of Sinentomon erythranum (Arthropoda: Hexapoda: Protura) underwent highly divergent evolution. BMC Evol. Biol. 2011, 11, 246–258. [Google Scholar] [CrossRef]
- Carapelli, A.; Liò, P.; Nardi, F.; Van Der Wath, E.; Frati, F. Phylogenetic analysis of mitochondrial protein coding genes confirms the reciprocal paraphyly of Hexapoda and Crustacea. BMC Evol. Biol. 2007, 7, S8. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Yin, W.Y.; Dallai, R.; Xue, L. Sperm evolution of Protura. In Proceedings of the 3rd International Seminar on Apterygota, Siena, Italy, 21–26 August 1989; Dallai, R., Ed.; pp. 195–198. [Google Scholar]
- Yin, W.Y. New considerations on systematics of Protura. In Proceedings of the XX International Congress of Entomology, Firenze, Italy, 25–31 August 1996; p. 60. [Google Scholar]
- Yin, W.Y.; Xie, R.D.; Yang, Y.M.; Yue, Q.Y.; Luan, Y.X. Analysis of the main characters for regrouping the class Protura (Hexapoda). Acta Zootaxonomic Sin. 2002, 27, 649–658. [Google Scholar]
- Yin, W.Y. The Protura, Fauna Sinica; Science Press: Beijing, China, 1999; pp. 1–510. [Google Scholar]
- Yin, W.Y. Studies on Chinese Protura I. Ten species of the genus Eosentomon from Nanking-Shanghai Regions. Acta Entomol. Sin. 1965, 14, 71–92. [Google Scholar]
- Tuxen, S.L. The genus Berberentulus (Insecta, Protura) with a key and phylogenetical considerations. Rev. Ecol. Biol. Sol. 1977, 14, 597–611. [Google Scholar]
- Imadaté, G. Taxonomic Arrangement of Japanese Protura (I); Bulletin of the National Science Museum: Tokyo, Japan, 1964; Volume 7, pp. 37–81. [Google Scholar]
- Xue, L.; Yin, W.Y. Further observations on the pseudoculi of Protura. In Proceedings of the 3rd International Seminar on Apterygota, Siena, Italy, 21–26 August 1989; Dallai, R., Ed.; pp. 253–263. [Google Scholar]
- Resch, M.C.; Shrubovych, J.; Bartel, D.; Szucsich, N.U.; Timelthaler, G.; Bu, Y.; Walzl, M.; Pass, G. Where taxonomy based on subtle morphological differences is perfectly mirrored by huge genetic distances: DNA Barcoding in Protura (Hexapoda). PLoS ONE 2014, 9, e90653. [Google Scholar] [CrossRef] [PubMed]
- Shrubovych, J.; Starý, J.; D’Haese, C.A. Molecular Phylogeny of Acerentomidae (Protura), with Description of Acerentuloides bernardi sp. nov. from North America. Fla. Entomol. 2017, 100, 433–443. [Google Scholar] [CrossRef]
- Qian, C.Y.; Bu, Y.; Luan, Y.X. DNA barcoding and an updated key to the genus Hesperentomon (Protura: Acerentomata: Hesperentomidae), with a new species from Northwest China. Zootaxa 2018, 4462, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Böhm, A.; Bartel, D.; Szucsich, N.U.; Pass, G. Confocal imaging of the exo- and endoskeleton of Protura after non- destructive DNA extraction. Soil Org. 2011, 83, 335–345. [Google Scholar]
- Gao, Y.; Bu, Y.; Luan, Y.X. Phylogenetic relationships of basal hexapods reconstructed from nearly complete 18S and 28S rRNA gene sequences. Zool. Sci. 2008, 25, 1139–1145. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, R.; Pedersen, A.G. RevTrans—Constructing alignments of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003, 31, 3537–3539. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Nylander, J. MrModeltest V2. Program distributed by the author. Bioinformatics 2004, 24, 581–583. [Google Scholar] [CrossRef]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. 2014. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 2 July 2018).
- Mesquite 3.51. Available online: https://github.com/MesquiteProject/MesquiteCore (accessed on 15 June 2018).
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Zhang, J.J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Barraclough, T.G. Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Brower, A.V.Z. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial-DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6491–6495. [Google Scholar] [CrossRef] [PubMed]
- Ezard, T.; Fujisawa, T.; Barraclough, T. Splits: Species’ Limits by Threshold Statistics. R Package Version 1.0-19. 2014. Available online: http://R-Forge.R-project.org/projects/splits/ (accessed on 25 May 2018).
- Yang, Z.; Rannala, B. Unguided species delimitation using DNA sequence data from multiple loci. Mol. Biol. Evol. 2014, 31, 3125–3135. [Google Scholar] [CrossRef]
- Nardi, F.; Carapelli, A.; Frati, F. Repeated regions in mitochondrial genomes: Distribution, origin and evolutionary significance. Mitochondrion 2012, 12, 483–491. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. The complete mitochondrial DNA sequence of the horseshoe crab Limulus polyphemus. Mol. Biol. Evol. 2000, 17, 813–824. [Google Scholar] [CrossRef]
- Comandi, S.; Carapelli, A.; Podsiadlowski, L.; Nardi, F.; Frati, F. The complete mitochondrial genome of Atelura formicaria (Hexapoda: Zygentoma) and the phylogenetic relationships of basal insects. Gene 2009, 439, 25–34. [Google Scholar] [CrossRef]
- Torricelli, G.; Carapelli, A.; Convey, P.; Nardi, F.; Boore, J.L.; Frati, F. High divergence across the whole mitochondrial genome in the “pan-Antarctic” springtail Friesea grisea: Evidence for cryptic species? Gene 2010, 449, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R.; Okimoto, R.; Macfarlane, J.L. Nucleotide correlations that suggest tertiary interactions in the TV-Replacement loop-containing mitochondrial transfer-RNAs of the nematodes. Nucleic Acids Res. 1994, 22, 4300–4306. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sun, H.; Guo, H.; Pan, D.; Qian, C.; Hao, S.; Zhou, K. The complete mitochondrial genome of Pauropus longiramus (Myriapoda: Pauropoda): Implications on early diversification of the myriapods revealed from comparative analysis. Gene 2012, 505, 57–65. [Google Scholar] [CrossRef]
- Xue, X.F.; Guo, J.F.; Dong, Y.; Hong, X.Y.; Shaob, R. Mitochondrial genome evolution and tRNA truncation in Acariformes mites: New evidence from eriophyoid mites. Sci. Rep. 2016, 6, 18920. [Google Scholar] [CrossRef]
- Podsiadlowski, L.; Carapelli, A.; Nardi, F.; Dallai, R.; Koch, M.; Frati, F.; Boore, J.L.F. The mitochondrial genomes of Campodea fragilis and Campodea lubbocki (Hexapoda: Diplura): High genetic divergence in a morphologically uniform taxon. Gene 2006, 381, 49–61. [Google Scholar] [CrossRef]
- Carapelli, A.; Torricelli, G.; Nardi, F.; Frati, F. The complete mitochondrial genome of the Antarctic sea spider Ammothea carolinensis (Chelicerata; Pycnogonida). Polar Biol. 2013, 36, 593–602. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Xue, L.; Dallai, R.; Yin, W.Y. Comparative tracheal structures of Apterygota. Acta Zool. Fenn. 1994, 195, 143–149. [Google Scholar]
- Yin, W.Y. New idea on phylogeny of Protura. In Proceedings of the XVII International Congress of Entomology, Hamburg, FDR, 20–26 August 1984; p. 7. [Google Scholar]
- Baccetti, B. Ultrastructure of sperm and its bearing on arthropod phylogeny. In Arthopod Phylogeny; Gupta, A.P., Ed.; Van Nostrand Reinhold Co.: New York, NY, USA, 1979; pp. 609–644. [Google Scholar]
- Bu, Y.; Ma, Y.; Luan, Y.X. Paracerella Imadate in China: The description of a new species and the analysis of genetic differences between populations (Protura, Acerentomata, Nipponentomidae). ZooKeys 2016, 604, 1–11. [Google Scholar] [CrossRef][Green Version]
- Sukurman, J.; Knowles, L.L. Multispecies coalescent delimits structure, not species. Proc. Natl. Acad. Sci. USA 2017, 114, 1607–1612. [Google Scholar] [CrossRef]
- Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–417. [Google Scholar] [CrossRef]
- Schlick-Steiner, B.C.; Steiner, F.M.; Seifert, B.; Stauffer, C.; Christian, E.; Crozier, R.H. Integrative taxonomy: A multisource approach to exploring biodiversity. Annu. Rev. Entomol. 2010, 55, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to fail at species delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | A | C | G | T | Length (bp) | Positions | Intergenic Spacers/Overlaps | Strand | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|---|---|---|
| Luag | 51.61 | 3.23 | 6.45 | 38.71 | 62 | 416–477 | 8 | J | ||
| nad2 | 34.91 | 8.49 | 7.02 | 49.58 | 954 | 486–1439 | 4 | J | I | TAA |
| trnD | 48.39 | 1.61 | 6.45 | 43.55 | 62 | 1444–1505 | 6 | J | ||
| nad3 | 32.20 | 8.19 | 11.02 | 48.59 | 354 | 1512–1865 | 70 | J | F | TAA |
| trnT | 42.19 | 3.13 | 10.94 | 43.75 | 64 | 1936–1999 | −1 | J | ||
| trnE | 40.32 | 4.84 | 8.07 | 46.77 | 62 | 1999–2060 | 15 | J | ||
| nad6 | 36.07 | 6.97 | 8.46 | 48.51 | 402 | 2076–2477 | 4 | J | I | TAA |
| cob | 32.95 | 11.37 | 10.48 | 45.20 | 1126 | 2482–3607 | 0 | J | M | T |
| trnSuga | 42.86 | 4.76 | 9.52 | 42.86 | 63 | 3608–3670 | 8 | J | ||
| nad5 | 34.90 | 8.60 | 9.27 | 47.23 | 1662 | 3679–5340 | 6 | N | L | TAA |
| trnH | 37.29 | 3.39 | 13.56 | 45.76 | 59 | 5347–5405 | −2 | N | ||
| trnG | 45.00 | 5.00 | 8.33 | 41.67 | 60 | 5404–5463 | −3 | J | ||
| nad1 | 33.95 | 8.54 | 11.01 | 46.50 | 972 | 5461–6432 | −16 | N | I | TAA |
| trnLuaa | 50.77 | 0.00 | 0.00 | 49.23 | 65 | 6417–6481 | 0 | J | ||
| rrnS | 43.11 | 7.62 | 10.05 | 39.22 | 617 | 6482–7098 | 0 | J | ||
| trnQ | 37.50 | 3.13 | 7.81 | 51.56 | 64 | 7099–7162 | −3 | N | ||
| trnM | 40.91 | 13.64 | 12.12 | 33.33 | 66 | 7160–7225 | 2 | J | ||
| trnP | 40.32 | 4.84 | 8.07 | 46.77 | 62 | 7228–7289 | −1 | J | ||
| trnC | 51.85 | 1.85 | 3.70 | 42.59 | 54 | 7289–7342 | 13 | N | ||
| trnW | 47.76 | 7.46 | 5.97 | 38.81 | 67 | 7356–7422 | −3 | J | ||
| trnY | 36.92 | 9.23 | 15.39 | 38.46 | 65 | 7420–7484 | 5 | N | ||
| cox1 | 31.14 | 13.26 | 13.52 | 42.07 | 1538 | 7490–9027 | 0 | J | M | TA |
| cox2 | 37.63 | 11.09 | 9.15 | 42.13 | 667 | 9028–9694 | 5 | J | M | T |
| trnK | 34.48 | 15.52 | 12.07 | 37.93 | 58 | 9700–9757 | 7 | J | ||
| atp8 | 49.36 | 7.69 | 1.92 | 41.03 | 156 | 9765–9920 | −1 | J | I | TAA |
| atp6 | 35.28 | 9.87 | 8.37 | 46.49 | 669 | 9920–10588 | −1 | J | I | TAA |
| cox3 | 33.21 | 11.11 | 10.99 | 44.70 | 792 | 10588–11379 | 2 | J | M | TAA |
| trnN | 47.62 | 3.18 | 7.94 | 41.27 | 63 | 11382–11444 | −1 | J | ||
| trnV | 48.28 | 10.35 | 8.62 | 32.76 | 58 | 11444–11501 | 60 | J | ||
| trnR | 35.82 | 13.43 | 11.94 | 38.81 | 67 | 11562–11628 | −25 | N | ||
| nad4L | 36.08 | 5.16 | 9.62 | 49.14 | 291 | 11604–11894 | −23 | J | M | TAA |
| nad4 | 36.34 | 9.46 | 9.69 | 44.52 | 1332 | 11872–13203 | 2 | J | I | TAA |
| trnF | 44.62 | 4.62 | 10.77 | 40.00 | 65 | 13206–13270 | 0 | J | ||
| rrnL | 44.38 | 7.25 | 7.79 | 40.58 | 1104 | 13271–14374 | 0 | J | ||
| trnA | 49.23 | 4.62 | 9.23 | 36.92 | 65 | 14375–14439 | 298 | N | ||
| trnI | 34.43 | 8.20 | 18.03 | 39.34 | 61 | 14738–14798 | J | |||
| mean | 36.50 | 9.20 | 9.76 | 44.54 |
| Genes | A | C | G | T | Length (bp) | Positions | Intergenic Spacers/Overlaps | Strand | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|---|---|---|
| rrnS | 40.77 | 11.57 | 9.09 | 38.57 | 726 | 1–726 | 0 | N | ||
| trnLuaa | 49.18 | 4.92 | 3.28 | 42.62 | 61 | 727–787 | −2 | N | ||
| nad2 | 28.62 | 9.64 | 14.36 | 47.38 | 954 | 786–1739 | −3 | J | M | TAA |
| trnY | 37.10 | 8.07 | 16.13 | 38.71 | 62 | 1737–1798 | −1 | N | ||
| trnW | 39.71 | 10.29 | 8.82 | 41.18 | 68 | 1798–1865 | 1 | J | ||
| cox1 | 25.13 | 14.45 | 18.75 | 41.67 | 1536 | 1867–3402 | 4 | J | M | TAA |
| cox2 | 30.34 | 12.56 | 17.19 | 39.91 | 669 | 3407–4075 | 20 | J | M | TAA |
| trnK | 46.55 | 10.35 | 8.62 | 34.48 | 58 | 4096–4153 | −37 | J | ||
| trnD | 41.07 | 3.57 | 10.71 | 44.64 | 56 | 4117–4172 | 7 | J | ||
| atp8 | 33.33 | 9.52 | 9.52 | 47.62 | 147 | 4180–4326 | −13 | J | M | TAA |
| atp6 | 28.00 | 15.11 | 17.48 | 39.41 | 675 | 4314–4988 | −1 | J | M | TAA |
| cox3 | 27.29 | 13.71 | 17.59 | 41.41 | 722 | 4988–5779 | 12 | J | M | TAA |
| nad3 | 24.77 | 12.39 | 16.92 | 45.92 | 331 | 5792–6122 | 0 | J | M | T |
| trnA | 46.43 | 5.36 | 7.14 | 41.07 | 56 | 6123–6178 | 0 | J | ||
| trnT | 53.70 | 1.85 | 5.56 | 38.89 | 54 | 6179–6232 | 1 | J | ||
| trnSgcu | 46.55 | 8.62 | 6.90 | 37.93 | 58 | 6234–6291 | 1 | J | ||
| trnN | 49.12 | 8.77 | 5.26 | 36.84 | 57 | 6293–6349 | 28 | J | ||
| nad6 | 28.99 | 6.28 | 16.43 | 48.31 | 414 | 6378–6791 | −1 | J | M | TAA |
| cob | 26.04 | 13.20 | 17.36 | 43.40 | 1129 | 6791–7919 | 0 | J | M | T |
| trnSuga | 35.59 | 6.78 | 15.25 | 42.37 | 59 | 7920–7978 | −1 | J | ||
| trnF | 46.03 | 6.35 | 6.35 | 41.27 | 63 | 7978–8040 | −62 | N | ||
| trnE | 42.86 | 6.35 | 6.35 | 44.44 | 63 | 7979–8041 | −1 | J | ||
| nad5 | 32.47 | 15.48 | 11.91 | 40.15 | 1654 | 8041–9694 | 6 | N | M | T |
| nad4L | 33.93 | 8.57 | 10.71 | 46.79 | 280 | 9701–9980 | 5 | N | M | T |
| trnM | 36.92 | 15.39 | 12.31 | 35.39 | 65 | 9986–10050 | −2 | J | ||
| trnG | 40.00 | 16.67 | 15.00 | 28.33 | 60 | 10049–10108 | 0 | J | ||
| trnQ | 37.50 | 3.13 | 15.63 | 43.75 | 64 | 10109–10172 | 0 | J | ||
| trnH | 43.86 | 1.75 | 12.28 | 42.11 | 57 | 10173–10229 | 0 | N | ||
| nad4 | 29.86 | 15.77 | 14.33 | 40.03 | 1249 | 10230–11478 | 43 | N | M | T |
| trnLuag | 37.50 | 8.93 | 16.07 | 37.50 | 56 | 11522–11577 | 1 | N | ||
| trnV | 33.90 | 10.17 | 5.09 | 50.85 | 59 | 11579–11637 | −11 | N | ||
| trnR | 36.00 | 12.00 | 12.00 | 40.00 | 50 | 11627–11676 | −4 | J | ||
| trnC | 44.83 | 6.90 | 6.90 | 41.38 | 58 | 11673–11730 | 4 | N | ||
| nad1 | 25.78 | 14.02 | 21.25 | 38.94 | 927 | 11735–12661 | 1 | J | M | TAA |
| trnI | 46.77 | 8.07 | 11.29 | 33.87 | 62 | 12663–12724 | −2 | N | ||
| trnP | 40.00 | 3.33 | 13.33 | 43.33 | 60 | 12723–12782 | 0 | J | ||
| rrnL | 38.52 | 10.22 | 12.39 | 38.88 | 1106 | 12783–13888 | 0 | J | ||
| A+T-rich | 27.40 | 23.47 | 16.45 | 32.68 | 1325 | 13889–15213 | ||||
| mean | 30.85 | 13.49 | 15.01 | 40.65 |


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carapelli, A.; Bu, Y.; Chen, W.-J.; Nardi, F.; Leo, C.; Frati, F.; Luan, Y.-X. Going Deeper into High and Low Phylogenetic Relationships of Protura. Genes 2019, 10, 292. https://doi.org/10.3390/genes10040292
Carapelli A, Bu Y, Chen W-J, Nardi F, Leo C, Frati F, Luan Y-X. Going Deeper into High and Low Phylogenetic Relationships of Protura. Genes. 2019; 10(4):292. https://doi.org/10.3390/genes10040292
Chicago/Turabian StyleCarapelli, Antonio, Yun Bu, Wan-Jun Chen, Francesco Nardi, Chiara Leo, Francesco Frati, and Yun-Xia Luan. 2019. "Going Deeper into High and Low Phylogenetic Relationships of Protura" Genes 10, no. 4: 292. https://doi.org/10.3390/genes10040292
APA StyleCarapelli, A., Bu, Y., Chen, W.-J., Nardi, F., Leo, C., Frati, F., & Luan, Y.-X. (2019). Going Deeper into High and Low Phylogenetic Relationships of Protura. Genes, 10(4), 292. https://doi.org/10.3390/genes10040292