The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Antisense Transcription Start Sites Annotation
2.2. Antisense RNA Loci Inference
2.3. Promoter and 3′ End Sequence Analysis
2.4. Gene Functions
2.5. Type II Toxin-Antitoxin Systems Annotation and Antisense Transcription Start Sites Identification in Other Archaea
2.6. Differential Expression Analysis
2.7. Antisense Transcription Start Sites Comparison between Halobacterium salinarum and Haloferax volcanii
2.8. Ribo-Seq Data Analysis
3. Results
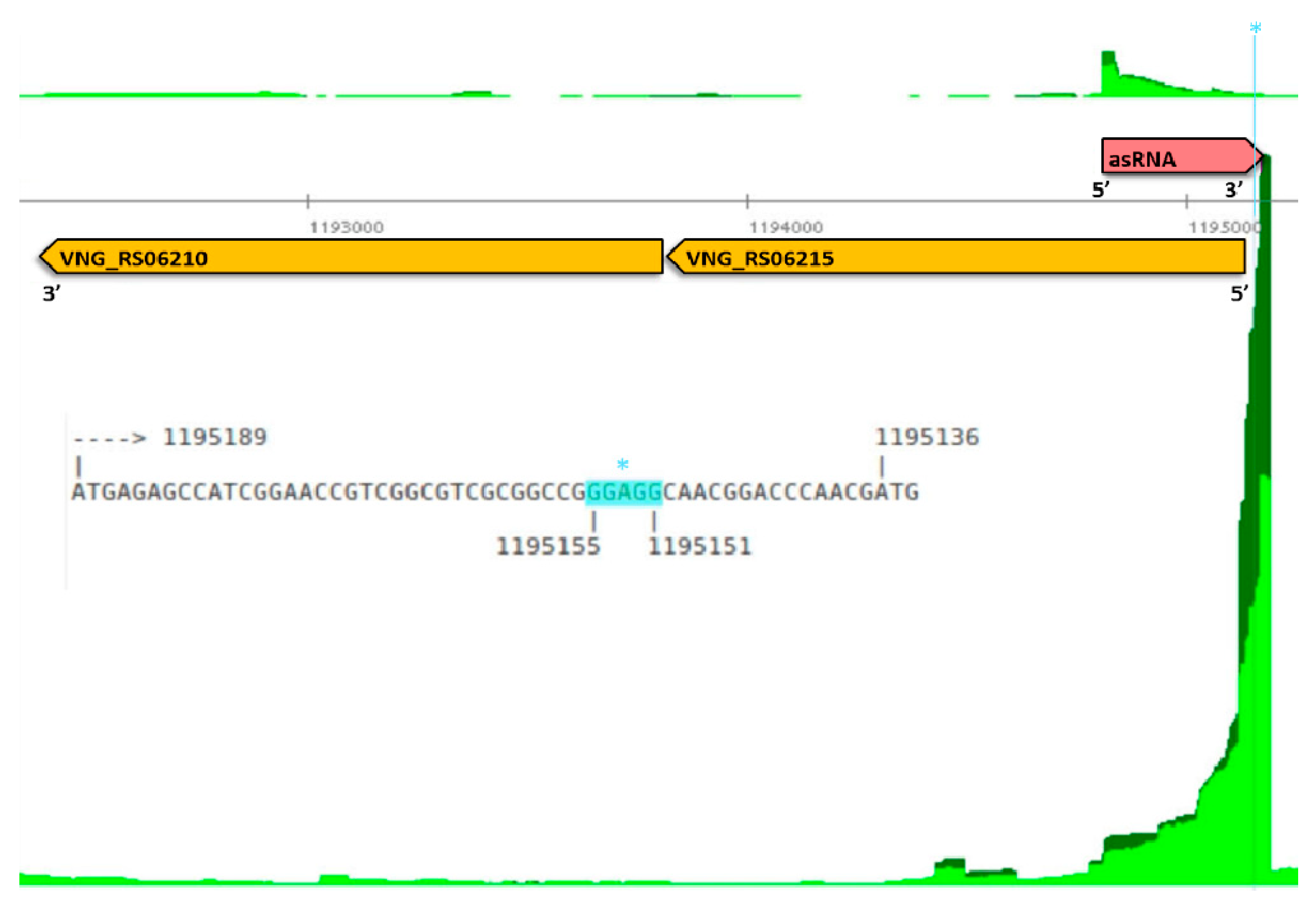
3.1. Mapping Primary Antisense RNAs
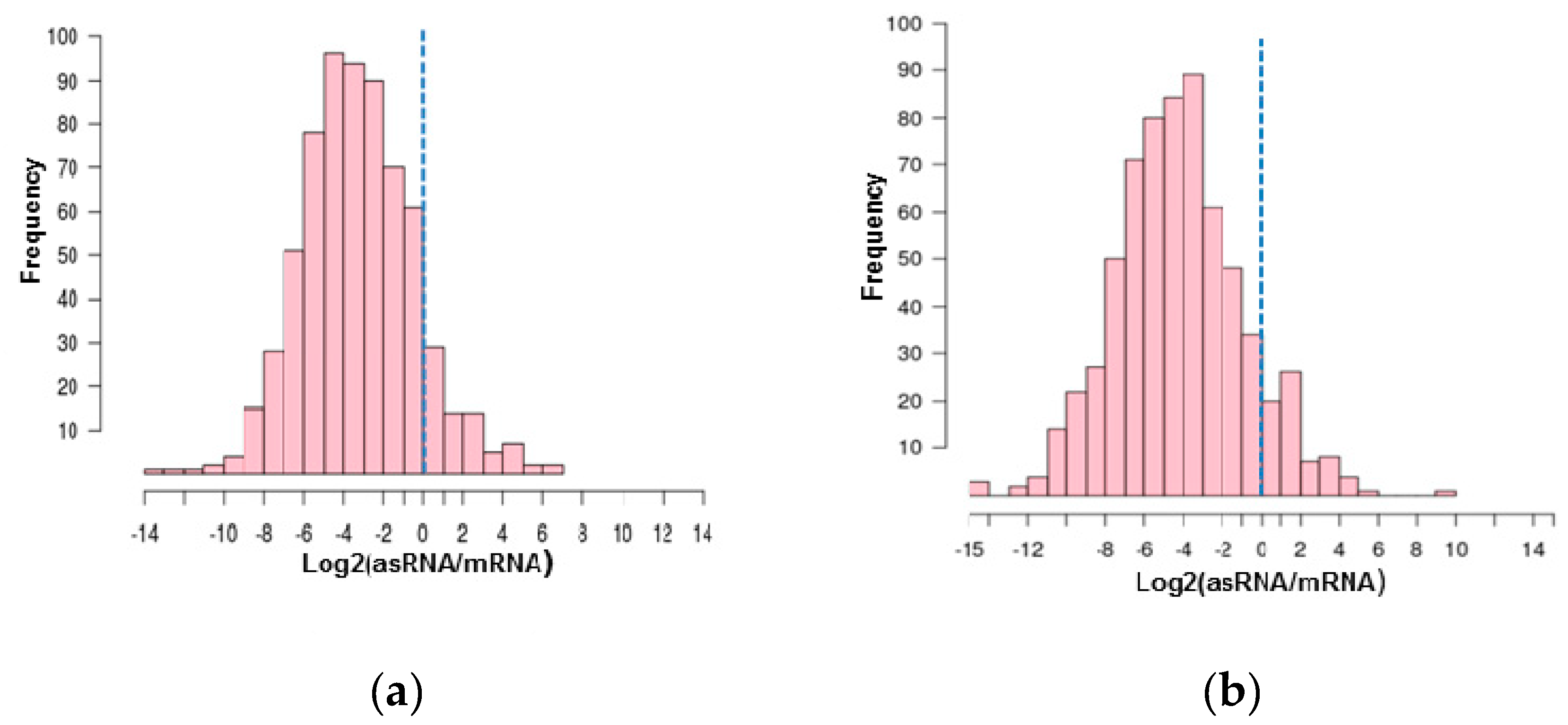
3.2. Antisense RNAs Expression Levels
3.3. Function of the Genes on the Opposite Strand of Antisense RNAs
3.4. Ribosome-Associated Antisense RNAs
3.5. Conservation of Antisense Transcription Start Sites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sayed, N.; Jousselin, A.; Felden, B. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat. Struct. Mol. Biol. 2012, 19, 105–113. [Google Scholar] [CrossRef]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef]
- Lasa, I.; Toledo-Arana, A.; Dobin, A.; Villanueva, M.; de Los Mozos, I.R.; Vergara-Irigaray, M.; Segura, V.; Fagegaltier, D.; Penadés, J.R.; Valle, J.; et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 20172–20177. [Google Scholar] [CrossRef]
- Kawano, M.; Aravind, L.; Storz, G. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 2007, 64, 738–754. [Google Scholar] [CrossRef]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–459. [Google Scholar] [CrossRef]
- Bøvre, K.; Szybalski, W. Patterns of convergent and overlapping transcription within the b2 region of coliphage λ. Virology 1969, 38, 614–626. [Google Scholar] [CrossRef]
- Inouye, M. Antisense RNA: Its functions and applications in gene regulation—A review. Gene 1988, 72, 25–34. [Google Scholar] [CrossRef]
- Vanhée-Brossollet, C.; Vaquero, C. Do natural antisense transcripts make sense in eukaryotes? Gene 1998, 211, 1–9. [Google Scholar] [CrossRef]
- Lasa, I.; Toledo-Arana, A.; Gingeras, T. An effort to make sense of antisense transcription in bacteria. RNA Biol. 2012, 9, 1039–1044. [Google Scholar] [CrossRef]
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar] [CrossRef]
- Sharma, C.M.; Vogel, J. Differential RNA-seq: The approach behind and the biological insight gained. Curr. Opin. Microbiol. 2014, 19, 97–105. [Google Scholar] [CrossRef]
- Sun, Y.; Li, D.; Zhang, R.; Peng, S.; Zhang, G.; Yang, T.; Qian, A. Strategies to identify natural antisense transcripts. Biochimie 2017, 132, 131–151. [Google Scholar] [CrossRef]
- Beiter, T.; Reich, E.; Williams, R.W.; Simon, P. Antisense transcription: A critical look in both directions. Cell. Mol. Life Sci. 2009, 66, 94–112. [Google Scholar] [CrossRef]
- Georg, J.; Hess, W.R. Cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 286–300. [Google Scholar] [CrossRef]
- Wade, J.T.; Grainger, D.C. Pervasive transcription: Illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014, 12, 647–653. [Google Scholar] [CrossRef]
- Lloréns-Rico, V.; Cano, J.; Kamminga, T.; Gil, R.; Latorre, A.; Chen, W.H.; Bork, P.; Glass, J.I.; Serrano, L.; Lluch-Senar, M. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2016, 2, e1501363. [Google Scholar] [CrossRef]
- Wagner, E.G.H.; Romby, P. Chapter three-small RNAs in bacteria and archaea: Who they are, what they do, and how they do it. Adv. Genet. 2015, 90, 133–208. [Google Scholar]
- Eckweiler, D.; Häussler, S. Antisense transcription in Pseudomonas aeruginosa. Microbiology 2018, 164, 889–895. [Google Scholar] [CrossRef]
- Krüger, K.; Pfeifer, F. Transcript analysis of the c-vac region and differential synthesis of the two regulatory gas vesicle proteins GvpD and GvpE in Halobacterium salinarium PHH4. J. Bacteriol. 1996, 178, 4012–4019. [Google Scholar] [CrossRef]
- Gelsinger, D.R.; DiRuggiero, J. The non-coding regulatory RNA revolution in archaea. Genes 2018, 9, 141. [Google Scholar] [CrossRef]
- Babski, J.; Haas, K.A.; Näther-Schindler, D.; Pfeiffer, F.; Förstner, K.U.; Hammelmann, M.; Hilker, R.; Becker, A.; Sharma, C.M.; Marchfelder, A.; et al. Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-Seq (dRNA-Seq). BMC Genom. 2016, 17, 629. [Google Scholar] [CrossRef]
- Li, J.; Qi, L.; Guo, Y.; Yue, L.; Li, Y.; Ge, W.; Wu, J.; Shi, W.; Dong, X. Global mapping transcriptional start sites revealed both transcriptional and post-transcriptional regulation of cold adaptation in the methanogenic archaeon Methanolobus psychrophilus. Sci. Rep. 2015, 5, 9209. [Google Scholar] [CrossRef] [PubMed]
- Jäger, D.; Förstner, K.U.; Sharma, C.M.; Santangelo, T.J.; Reeve, J.N. Primary transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis. BMC Genom. 2014, 15, 684. [Google Scholar] [CrossRef] [PubMed]
- Jäger, D.; Sharma, C.M.; Thomsen, J.; Ehlers, C.; Vogel, J.; Schmitz, R.A. Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc. Natl. Acad. Sci. USA 2009, 106, 21878–21882. [Google Scholar] [CrossRef]
- Cho, S.; Kim, M.-S.; Jeong, Y.; Lee, B.-R.; Lee, J.-H.; Kang, S.G.; Cho, B.-K. Genome-wide primary transcriptome analysis of H2-producing archaeon Thermococcus onnurineus NA1. Sci. Rep. 2017, 7, 43044. [Google Scholar] [CrossRef]
- Smollett, K.; Blombach, F.; Reichelt, R.; Thomm, M.; Werner, F. A global analysis of transcription reveals two modes of Spt4/5 recruitment to archaeal RNA polymerase. Nat. Microbiol. 2017, 2, 17021. [Google Scholar] [CrossRef] [PubMed]
- Gelsinger, D.R.; DiRuggiero, J. Transcriptional landscape and regulatory roles of small noncoding RNAs in the oxidative stress response of the Haloarchaeon Haloferax volcanii. J. Bacteriol. 2018, 200, e00779-17. [Google Scholar] [CrossRef] [PubMed]
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef]
- Oesterhelt, D.; Stoeckenius, W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat. New Biol. 1971, 233, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Trüper, H. Anaerobic growth of halophilic archaeobacteria by reduction of dimethysulfoxide and trimethylamine N-oxide. FEMS Microbiol. Lett. 1990, 70, 33–36. [Google Scholar] [CrossRef]
- Ruepp, A.; Soppa, J. Fermentative arginine degradation in Halobacterium salinarium (Formerly Halobacterium halobium): Genes, gene products, and transcripts of the arcRACB gene cluster. J. Bacteriol. 1996, 178, 4942–4947. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Pan, M.; Meislin, M.; Facciotti, M.T.; El-Gewely, R.; Baliga, N.S. A systems view of haloarchaeal strategies to withstand stress from transition metals. Genome Res. 2006, 16, 841–854. [Google Scholar] [CrossRef]
- Coker, J.A.; DasSarma, P.; Kumar, J.; Müller, J.A.; DasSarma, S. Transcriptional profiling of the model Archaeon Halobacterium sp. NRC-1: Responses to changes in salinity and temperature. Saline Syst. 2007, 3, 6. [Google Scholar] [CrossRef]
- Baliga, N.S.; Bjork, S.J.; Bonneau, R.; Pan, M.; Iloanusi, C.; Kottemann, M.C.H.; Hood, L.; DiRuggiero, J. Systems level insights into the stress response to UV radiation in the halophilic archaeon Halobacterium NRC-1. Genome Res. 2004, 14, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, R.; Facciotti, M.T.; Reiss, D.J.; Schmid, A.K.; Pan, M.; Kaur, A.; Thorsson, V.; Shannon, P.; Johnson, M.H.; Bare, J.C.; et al. A predictive model for transcriptional control of physiology in a free living cell. Cell 2007, 131, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Reiss, D.J.; Allard, A.; Wu, W.-J.; Salvanha, D.M.; Plaisier, C.L.; Chandrasekaran, S.; Pan, M.; Kaur, A.; Baliga, N.S. A system-level model for the microbial regulatory genome. Mol. Syst. Biol. 2014, 10, 740. [Google Scholar] [CrossRef]
- Koide, T.; Reiss, D.J.; Bare, J.C.; Pang, W.L.; Facciotti, M.T.; Schmid, A.K.; Pan, M.; Marzolf, B.; Van, P.T.; Lo, F.Y.; et al. Prevalence of transcription promoters within archaeal operons and coding sequences. Mol. Syst. Biol. 2009, 5, 285. [Google Scholar] [CrossRef]
- Ten-Caten, F.; Vêncio, R.Z.N.; Lorenzetti, A.P.R.; Zaramela, L.S.; Santana, A.C.; Koide, T. Internal RNAs overlapping coding sequences can drive the production of alternative proteins in archaea. RNA Biol. 2018, 15, 1119–1132. [Google Scholar] [CrossRef]
- Zaramela, L.S.; Vêncio, R.Z.N.; Ten-Caten, F.; Baliga, N.S.; Koide, T. Transcription start site associated RNAs (TSSaRNAs) are ubiquitous in all domains of life. PLoS ONE 2014, 9, e107680. [Google Scholar] [CrossRef]
- Gomes-Filho, J.V.; Zaramela, L.S.; da Silva Italiani, V.C.; Baliga, N.S.; Vêncio, R.Z.N.; Koide, T. Sense overlapping transcripts in IS1341-type transposase genes are functional non-coding RNAs in archaea. RNA Biol. 2015, 12, 490–500. [Google Scholar] [CrossRef]
- Stolt, P.; Zillig, W. Structure specific ds/ss-RNase activity in the extreme halophile Halobacterium salinarium. Nucleic Acids Res. 1993, 21, 5595–5599. [Google Scholar] [CrossRef]
- Wagner, E.G.H.; Simons, R.W. Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 1994, 48, 713–742. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kahles, A.; Behr, J.; Rätsch, G. MMR: A tool for read multi-mapper resolution. Bioinformatics 2015, 32, 770–772. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Bare, J.C.; Koide, T.; Reiss, D.J.; Tenenbaum, D.; Baliga, N.S. Integration and visualization of systems biology data in context of the genome. BMC Bioinform. 2010, 11, 382. [Google Scholar] [CrossRef]
- Amman, F.; Wolfinger, M.T.; Lorenz, R.; Hofacker, I.L.; Stadler, P.F.; Findeiß, S. TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinform. 2014, 15, 89. [Google Scholar] [CrossRef]
- Pfeiffer, F.; Oesterhelt, D. A manual curation strategy to improve genome annotation: Application to a set of Haloarchael genomes. Life 2015, 5, 1427–1444. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Dehal, P.S.; Joachimiak, M.P.; Price, M.N.; Bates, J.T.; Baumohl, J.K.; Chivian, D.; Friedland, G.D.; Huang, K.H.; Keller, K.; Novichkov, P.S.; et al. MicrobesOnline: An integrated portal for comparative and functional genomics. Nucleic Acids Res. 2009, 38, D396–D400. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016, 45, D200–D203. [Google Scholar] [CrossRef]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.-Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef]
- Thomason, M.K.; Bischler, T.; Eisenbart, S.K.; Förstner, K.U.; Zhang, A.; Herbig, A.; Nieselt, K.; Sharma, C.M.; Storz, G. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J. Bacteriol. 2015, 197, 18–28. [Google Scholar] [CrossRef]
- Lybecker, M.; Zimmermann, B.; Bilusic, I.; Tukhtubaeva, N.; Schroeder, R. The double-stranded transcriptome of Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, 3134–3139. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, M.D.; Winsor, G.L.; Laird, M.R.; Brinkman, F.S.L. OrtholugeDB: A bacterial and archaeal orthology resource for improved comparative genomic analysis. Nucleic Acids Res. 2012, 41, D366–D376. [Google Scholar] [CrossRef] [PubMed]
- Babski, J.; Maier, L.-K.; Heyer, R.; Jaschinski, K.; Prasse, D.; Jäger, D.; Randau, L.; Schmitz, R.A.; Marchfelder, A.; Soppa, J. Small regulatory RNAs in Archaea. RNA Biol. 2014, 11, 484–493. [Google Scholar] [CrossRef]
- Kim, D.; Hong, J.S.-J.; Qiu, Y.; Nagarajan, H.; Seo, J.H.; Cho, B.K.; Tsai, S.F.; Palsson, B.Ø. Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 2012, 8, e1002867. [Google Scholar] [CrossRef]
- Wurtzel, O.; Sapra, R.; Chen, F.; Zhu, Y.; Simmons, B.A.; Sorek, R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010, 20, 133–141. [Google Scholar] [CrossRef]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.M.; Taylor, M.S.; Engström, P.G.; Frith, M.C.; et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef]
- Bell, S.D.; Jackson, S.P. Mechanism and regulation of transcription in archaea. Curr. Opin. Microbiol. 2001, 4, 208–213. [Google Scholar] [CrossRef]
- Seitzer, P.; Wilbanks, E.G.; Larsen, D.J.; Facciotti, M.T. A Monte Carlo-based framework enhances the discovery and interpretation of regulatory sequence motifs. BMC Bioinform. 2012, 13, 317. [Google Scholar] [CrossRef]
- Dar, D.; Prasse, D.; Schmitz, R.A.; Sorek, R. Widespread formation of alternative 3′ UTR isoforms via transcription termination in archaea. Nat. Microbiol. 2016, 1, 16143. [Google Scholar] [CrossRef]
- Brenneis, M.; Hering, O.; Lange, C.; Soppa, J. Experimental characterization of cis-acting elements important for translation and transcription in halophilic archaea. PLoS Genet. 2007, 3, e229. [Google Scholar] [CrossRef]
- Bouvier, M.; Sharma, C.M.; Mika, F.; Nierhaus, K.H.; Vogel, J. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol. Cell 2008, 32, 827–837. [Google Scholar] [CrossRef]
- Fozo, E.M.; Hemm, M.R.; Storz, G. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 2008, 72, 579–589. [Google Scholar] [CrossRef]
- Pfeifer, F.; Krüger, K.; Röder, R.; Mayr, A.; Ziesche, S.; Offner, S. Gas vesicle formation in halophilic Archaea. Arch. Microbiol. 1997, 167, 259–268. [Google Scholar] [CrossRef]
- Csiszàr, K.; Houmard, J.; Damerval, T.; de Marsac, N.T. Transcriptional analysis of the cyanobacterial gvpABC operon in differentiated cells: Occurrence of an antisense RNA complementary to three overlapping transcripts. Gene 1987, 60, 29–37. [Google Scholar] [CrossRef]
- Tarasov, V.; Schwaiger, R.; Furtwängler, K.; Dyall-Smith, M.; Oesterhelt, D. A small basic protein from the brz-brb operon is involved in regulation of bop transcription in Halobacterium salinarum. BMC Mol. Biol. 2011, 12, 42. [Google Scholar] [CrossRef]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, targets, and triggers: An overview of toxin-antitoxin biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef]
- Tang, T.H.; Polacek, N.; Zywicki, M.; Huber, H.; Brugger, K.; Garrett, R.; Bachellerie, J.P.; Hüttenhofer, A. Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus. Mol. Microbiol. 2005, 55, 469–481. [Google Scholar] [CrossRef]
- Ellis, M.J.; Trussler, R.S.; Haniford, D.B. A cis-encoded sRNA, Hfq and mRNA secondary structure act independently to suppress IS200 transposition. Nucleic Acids Res. 2015, 43, 6511–6527. [Google Scholar] [CrossRef]
- Pircher, A.; Gebetsberger, J.; Polacek, N. Ribosome-associated ncRNAs: An emerging class of translation regulators. RNA Biol. 2014, 11, 1335–1339. [Google Scholar] [CrossRef]
- Wyss, L.; Waser, M.; Gebetsberger, J.; Zywicki, M.; Polacek, N. mRNA-specific translation regulation by a ribosome-associated ncRNA in Haloferax volcanii. Sci. Rep. 2018, 8, 12502. [Google Scholar] [CrossRef]
- Raghavan, R.; Sloan, D.B.; Ochman, H.C. Antisense transcription is pervasive but rarely conserved in enteric bacteria. MBio 2012, 3, e00156-12. [Google Scholar] [CrossRef]
- Dugar, G.; Herbig, A.; Förstner, K.U.; Heidrich, N.; Reinhardt, R.; Nieselt, K.; Sharma, C.M. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 2013, 9, e1003495. [Google Scholar] [CrossRef]
- Shao, W.; Price, M.N.; Deutschbauer, A.M.; Romine, M.F.; Arkin, A.P. Conservation of transcription start sites within genes across a bacterial genus. MBio 2014, 5, e01398-14. [Google Scholar] [CrossRef]
- Kopf, M.; Klähn, S.; Scholz, I.; Hess, W.R.; Voß, B. Variations in the non-coding transcriptome as a driver of inter-strain divergence and physiological adaptation in bacteria. Sci. Rep. 2015, 5, 9560. [Google Scholar] [CrossRef]
- Mei, Y.; Liu, H.; Zhang, S.; Yang, M.; Hu, C.; Zhang, J.; Shen, P.; Chen, X. Effects of salinity on the cellular physiological responses of Natrinema sp. J7-2. PLoS ONE 2017, 12, e0184974. [Google Scholar] [CrossRef]
- Irnov, I.; Sharma, C.M.; Vogel, J.; Winkler, W.C. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010, 38, 6637–6651. [Google Scholar] [CrossRef]
- Van der Meulen, S.B.; de Jong, A.; Kok, J. Transcriptome landscape of Lactococcus lactis reveals many novel RNAs including a small regulatory RNA involved in carbon uptake and metabolism. RNA Biol. 2016, 13, 353–366. [Google Scholar] [CrossRef][Green Version]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469. [Google Scholar] [CrossRef]
- Santangelo, T.J.; Reeve, J.N. Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J. Mol. Biol. 2006, 355, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Brosius, J.; Gould, S.J. On “genomenclature”: A comprehensive (and respectful) taxonomy for pseudogenes and other “junk DNA”. Proc. Natl. Acad. Sci. USA 1992, 89, 10706–10710. [Google Scholar] [CrossRef]
- Goyal, A.; Fiškin, E.; Gutschner, T.; Polycarpou-Schwarz, M.; Groß, M.; Neugebauer, J.; Gandhi, M.; Caudron-Herger, M.; Benes, V.; Diederichs, S. A cautionary tale of sense-antisense gene pairs: Independent regulation despite inverse correlation of expression. Nucleic Acids Res. 2017, 45, 12496–12508. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aTSS ID | asRNA ID | Strand | Start | End | Size (nt) | Locus | Annotation |
|---|---|---|---|---|---|---|---|
| aTSS_1555 | VNG_as12280_1555 | + | 16743 | 16863 | 120 | VNG_RS12280 | gvpL |
| aTSS_1556 | VNG_as12280_1556 | + | 17055 | 17138 | 83 | VNG_RS12280 | gvpL |
| aTSS_1557 | VNG_as12290_1557 | + | 18092 | 18168 | 76 | VNG_RS12290 | gvpJ |
| aTSS_1558 | VNG_as13760_1558 | + | 18390 | 18428 | 38 | VNG_RS13760 | gvpI |
| aTSS_1559 | VNG_as13760_1559 | + | 18615 | 18643 | 28 | VNG_RS13760 | gvpI |
| aTSS_1560 | VNG_as12295_1560 | + | 18698 | 18722 | 24 | VNG_RS12295 | gvpH |
| aTSS_1565 | VNG_as12315_1565 | + | 21000 | 21165 | 165 | VNG_RS12315 | gvpD |
| aTSS_1567 | VNG_as12315_1567 | + | 22084 | 22106 | 22 | VNG_RS12315 | gvpD |
| aTSS_1568 | VNG_as12315_1568 | + | 22128 | 22292 | 164 | VNG_RS12315 | gvpD |
| aTSS_1569 | VNG_as12325_1569 | − | 22865 | 22964 | 99 | VNG_RS12325 | gvpC |
| aTSS_1570 | VNG_as12325_1570 | − | 23888 | 23940 | 52 | VNG_RS12325 | gvpC |
| aTSS ID | asRNA ID | Strand | Start | End | Size (nt) | Locus | Annotation |
|---|---|---|---|---|---|---|---|
| aTSS_175 | VNG_as00745_175 | + | 155806 | 155906 | 100 | VNG_RS00745 | halorhodopsin |
| *daTSS_36 | VNG_da3105F_36 | − | 1088797 | 1089100 | 303 | VNG_RS05710 VNG_OE3105F | brz—bacteriorhodopsin regulating zinc finger protein; brb—bacteriorhodopsin-regulating basic protein |
| aTSS_824 | VNG_as05715_824 | − | 1089545 | 1089615 | 70 | VNG_RS05715 | bacteriorhodopsin |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Almeida, J.P.P.; Vêncio, R.Z.N.; Lorenzetti, A.P.R.; ten-Caten, F.; Gomes-Filho, J.V.; Koide, T. The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1. Genes 2019, 10, 280. https://doi.org/10.3390/genes10040280
de Almeida JPP, Vêncio RZN, Lorenzetti APR, ten-Caten F, Gomes-Filho JV, Koide T. The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1. Genes. 2019; 10(4):280. https://doi.org/10.3390/genes10040280
Chicago/Turabian Stylede Almeida, João Paulo Pereira, Ricardo Z. N. Vêncio, Alan P. R. Lorenzetti, Felipe ten-Caten, José Vicente Gomes-Filho, and Tie Koide. 2019. "The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1" Genes 10, no. 4: 280. https://doi.org/10.3390/genes10040280
APA Stylede Almeida, J. P. P., Vêncio, R. Z. N., Lorenzetti, A. P. R., ten-Caten, F., Gomes-Filho, J. V., & Koide, T. (2019). The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1. Genes, 10(4), 280. https://doi.org/10.3390/genes10040280