DNA Methylation Patterns in the Social Spider, Stegodyphus dumicola



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Species
2.2. Sample Collections and Datasets
2.3. Whole Genome Sequencing, Assembly, and Annotation
2.3.1. DNA Extraction and Sequencing
2.3.2. Genome Assembly
2.3.3. Genome Annotation
2.4. Gene Expression
RNA Extraction and Sequencing
2.5. Whole Genome Bisulfite Sequencing
2.5.1. DNA Extraction and Sequencing
2.5.2. Mapping and Methylation Calling
2.6. Differential Gene Expression and Methylation of Lab Acclimated Spiders
2.7. DNA Methylation and Stability of Gene Expression
2.8. DNA Methylation in Two Social Stegodyphus Species
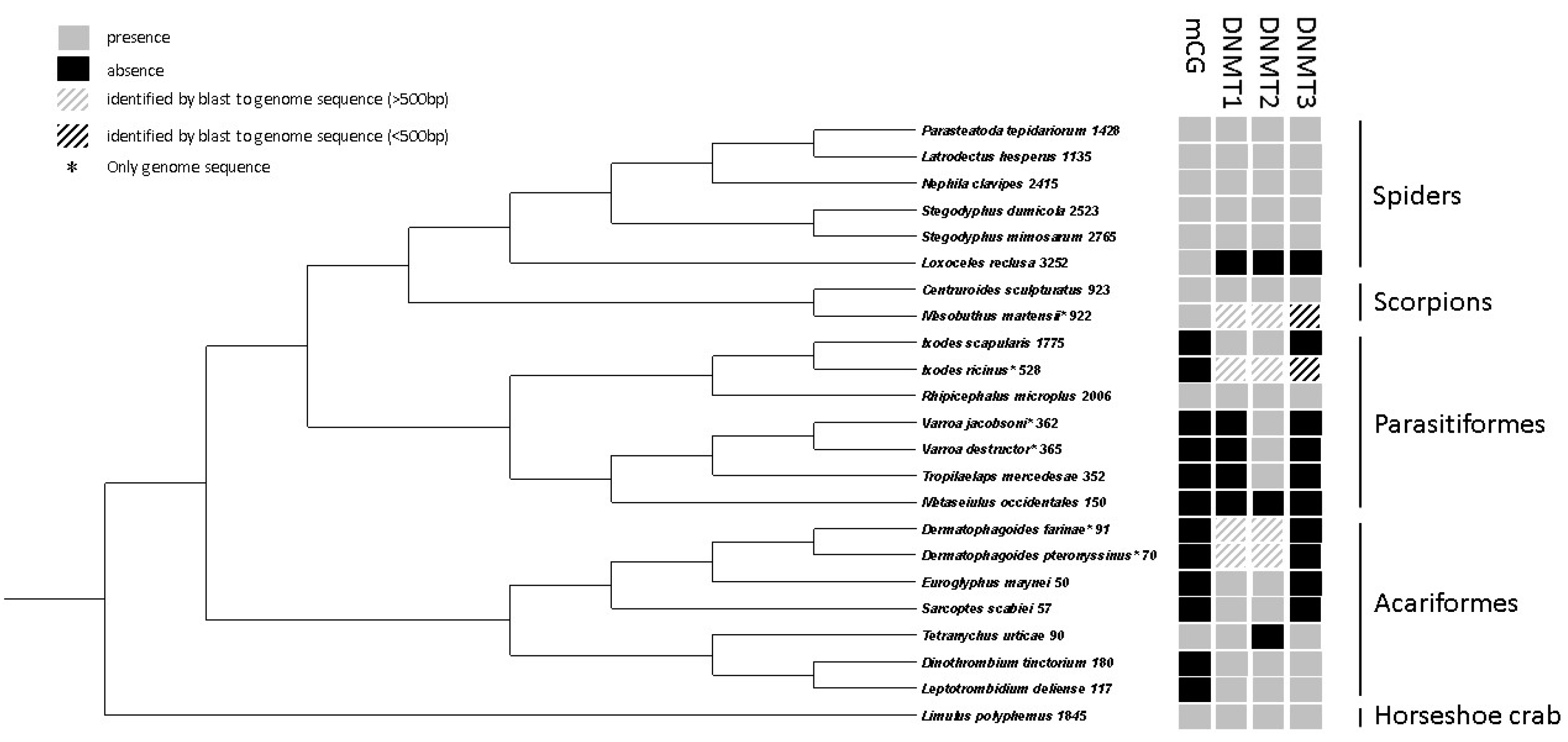
2.9. Comparative Analyses of DNA Methylation in Chelicerates
3. Results
3.1. Genome Assembly and Annotation
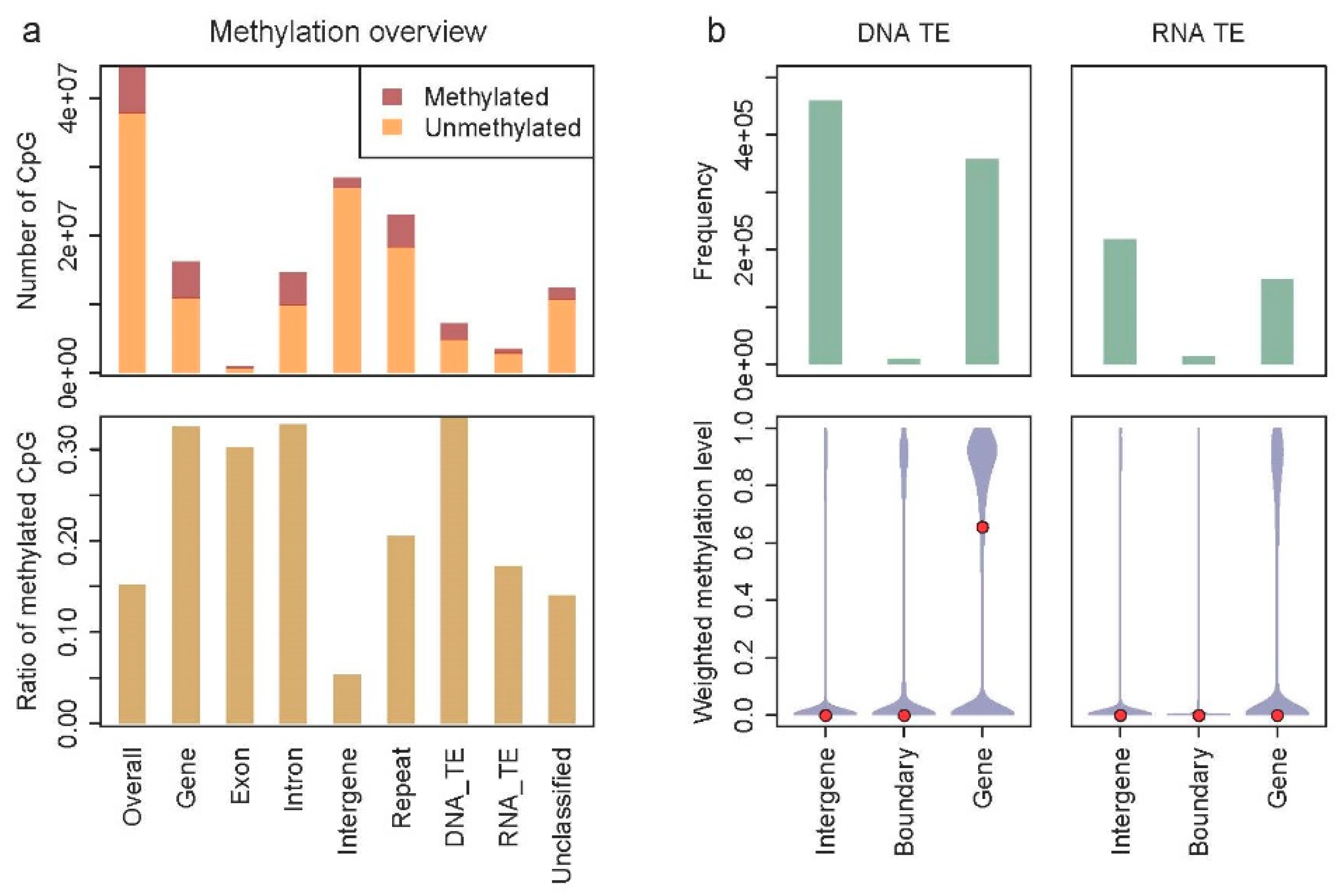
3.2. Methylation Pattern in Stegodyphus dumicola
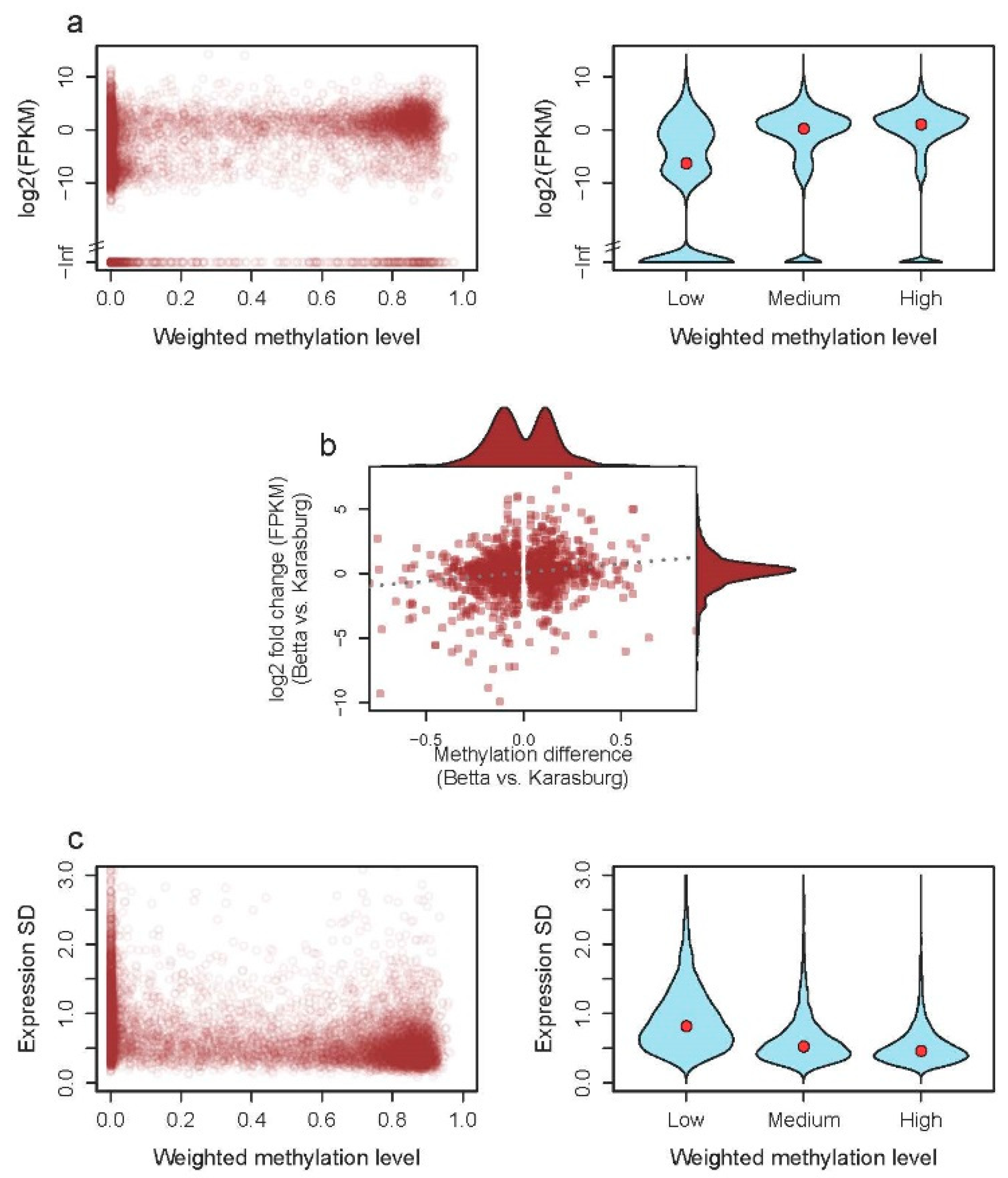
3.2.1. DNA Methylation and Gene Expression
3.2.2. DNA Methylation and Stability of Expression
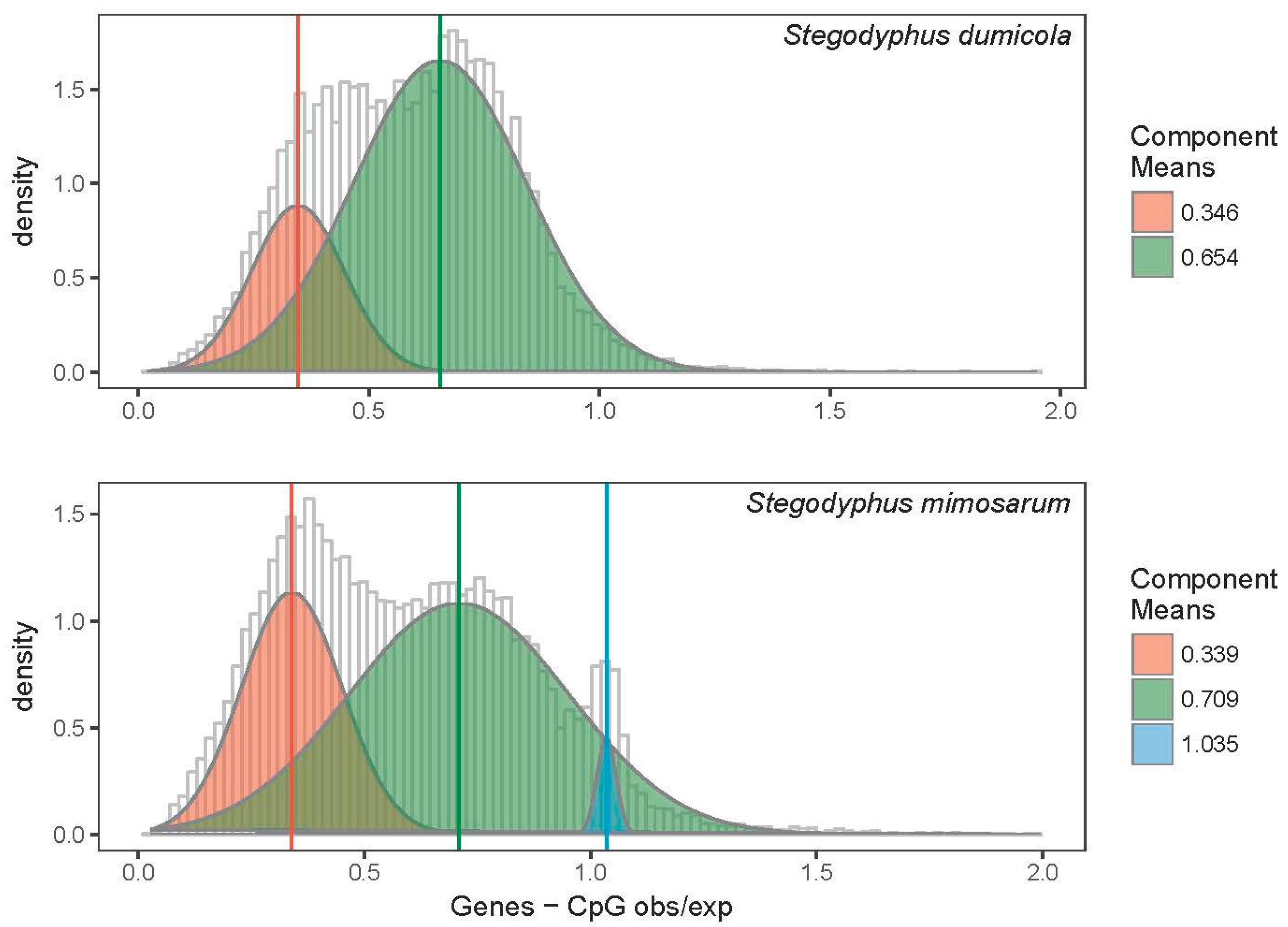
3.2.3. DNA Methylation in Two Social Stegodyphus Species
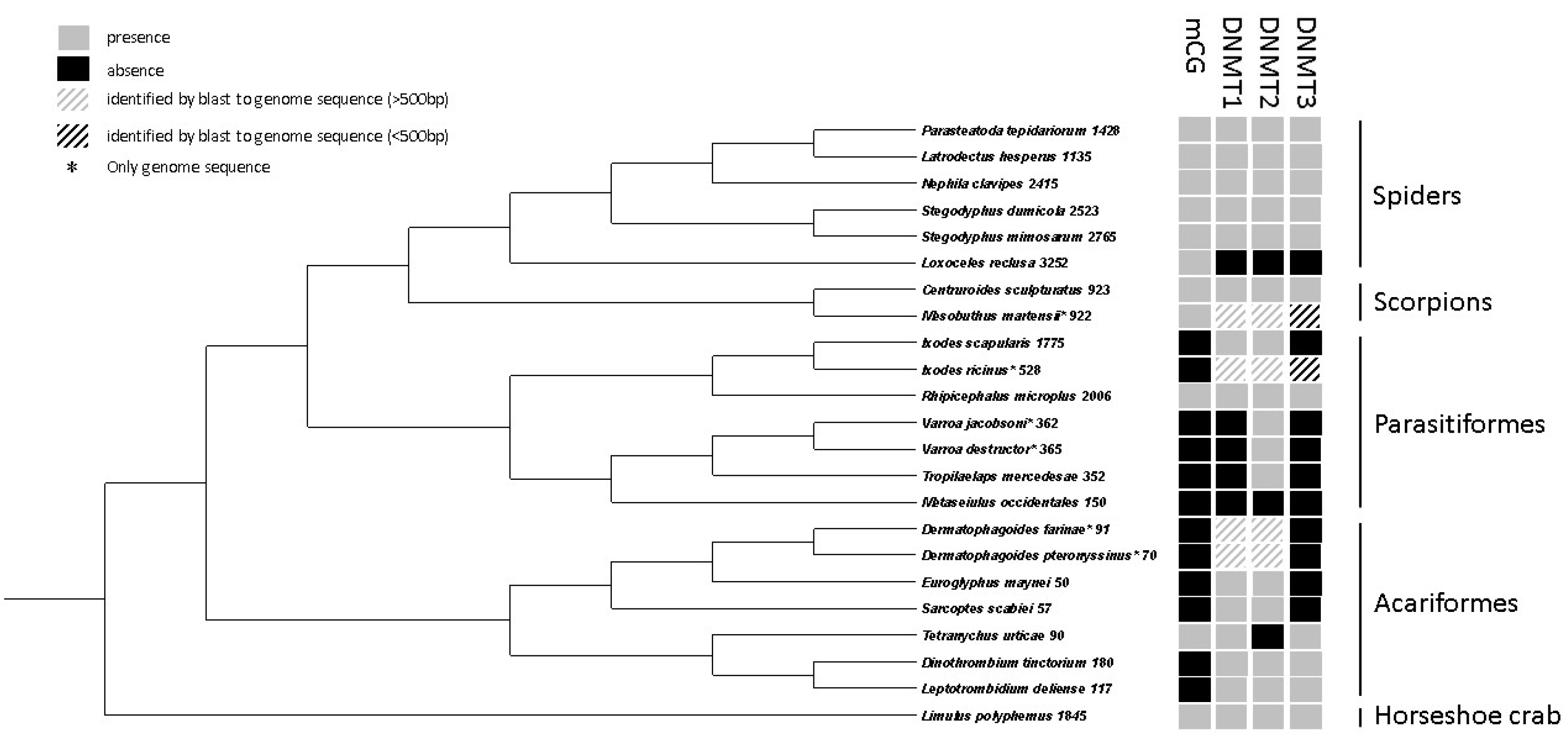
3.3. Comparative Analyses of DNA Methylation in Chelicerates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Data Accessibility
References
- Keller, T.E.; Han, P.; Yi, S.V. Evolutionary transition of promoter and gene body DNA methylation across invertebrate-vertebrate boundary. Mol. Biol. Evol. 2016, 33, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Varriale, A. DNA Methylation, Epigenetics, and evolution in vertebrates: Facts and challenges. Int. J. Evol. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Geiman, T.M.; Muegge, K. DNA methylation in early development. Mol. Reprod. Dev. 2010, 77, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Flores, K.B.; Wolschin, F.; Amdam, G.V. The role of methylation of DNA in environmental adaptation. Integr. Comp. Biol. 2013, 53, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Sarda, S.; Zeng, J.; Hunt, B.G.; Yi, S.V. The evolution of invertebrate gene body methylation. Mol. Biol. Evol. 2012, 29, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Kvist, J.; Athanasio, C.G.; Solari, O.S.; Brown, J.B.; Colbourne, J.K.; Pfrender, M.E.; Mirbahai, L. Pattern of DNA methylation in Daphnia: Evolutionary perspective. Genome Biol. Evol. 2018, 10, 1988–2007. [Google Scholar] [CrossRef] [PubMed]
- Gatzmann, F.; Falckenhayn, C.; Gutekunst, J.; Hanna, K.; Raddatz, G.; Carneiro, V.C.; Lyko, F.F. The methylome of the marbled crayfsh links gene body methylation to stable expression of poorly accessible genes. Epigenet. Chromatin 2018, 11, 57. [Google Scholar] [CrossRef]
- Bewick, A.J.; Vogel, K.J.; Moore, A.J.; Schmitz, R.J. Evolution of DNA methylation across Insects. Mol. Biol. Evol. 2017, 34, 654–665. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G.P. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuterd, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Beeler, S.M.; Wong, G.T.; Zheng, J.M.; Bush, E.C.; Remnant, E.J.; Oldroyd, B.P.; Drewell, R.A. Whole-genome DNA methylation profile of the Jewel Wasp (Nasonia vitripennis). G3-Genes Genomes Genet. 2014, 4, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Duret, L.; Mouchiroud, D. Determinants of substitution rates in mammalian genes: Expression pattern affects selection intensity but not mutation rate. Mol. Biol. Evol. 2000, 17, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Papp, B.; Hurst, L.D. Highly expressed genes in yeast evolve slowly. Genetics 2001, 158, 927–931. [Google Scholar] [PubMed]
- Takuno, S.; Gaut, B.S. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. USA 2013, 110, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Grbic, M.; Van Leeuwen, T.; Clark, R.M.; Rombauts, S.; Rouze, P.; Grbic, V.; Osborne, E.J.; Dermauw, W.; Phuong, C.T.N.; Ortego, F.; et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 2011, 479, 487–492. [Google Scholar] [CrossRef]
- Duncan, B.K.; Miller, J.H. Mutagenic deamination of cytosine residues in DNA. Nature 1980, 287, 560–561. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Mutagenesis at methylated CpG sequences. DNA Methylation Basic Mech. 2006, 301, 259–281. [Google Scholar]
- Settepani, V.; Bechsgaard, J.; Bilde, T. Phylogenetic analysis suggests that sociality is associated with reduced effectiveness of selection. Ecol. Evol. 2016, 6, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Lubin, Y.; Bilde, T. The evolution of sociality in spiders. Adv. Study Behav. 2007, 37, 83–145. [Google Scholar]
- Vanthournout, B.; Busck, M.M.; Bechsgaard, J.; Hendrickx, F.; Schramm, A.; Bilde, T. Male spiders control offspring sex ratio through greater production of female-determining sperm. Proc. R. Soc. B-Biol. Sci. 2018, 285. [Google Scholar] [CrossRef] [PubMed]
- Settepani, V.; Bechsgaard, J.; Bilde, T. Low genetic diversity and strong but shallow population differentiation suggests genetic homogenization by metapopulation dynamics in a social spider. J. Evol. Biol. 2014, 27, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- Settepani, V.; Schou, M.F.; Greve, M.; Grinsted, L.; Bechsgaard, J.; Bilde, T. Evolution of sociality in spiders leads to depleted genomic diversity at both population and species levels. Mol. Ecol. 2017, 26, 4197–4210. [Google Scholar] [CrossRef] [PubMed]
- Leffler, E.M.; Bullaughey, K.; Matute, D.R.; Meyer, W.K.; Segurel, L.; Venkat, A.; Andolfatto, P.; Przeworski, M. Revisiting an old riddle: What determines genetic diversity levels within species? PLoS Biol. 2012, 10, e1001388. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.X.; Hill, C.M.; Wu, S.G.; Ruan, J.; Ma, Z.S. DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef]
- Ye, C.X.; Ma, Z.S.S.; Cannon, C.H.; Pop, M.; Yu, D.W. Exploiting sparseness in de novo genome assembly. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.D.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 15 May 2018).
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.R.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, I351–I358. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://github.com/s-andrews/FastQC (accessed on 3 May 2018).
- Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 3 May 2018).
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357-U121. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Frazee, A.C.; Collado-Torres, L.; Jaffe, A.E.; Leek, J.T. Ballgown: Flexible, Isoform-Level Differential Expression Analysis; R Package Version 2.14.0; Bioconductor: Buffalo, NY, USA, 2018. [Google Scholar]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 10 January 2019).
- Benaglia, T.; Chauveau, D.; Hunter, D.R.; Young, D.S. mixtools: An R Package for analyzing finite mixture models. J. Stat. Softw. 2009, 32, 1–29. [Google Scholar] [CrossRef]
- Sanggaard, K.W.; Bechsgaard, J.S.; Fang, X.D.; Duan, J.J.; Dyrlund, T.F.; Gupta, V.; Jiang, X.T.; Cheng, L.; Fan, D.D.; Feng, Y.; et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 2014, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Loytynoja, A.; Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 2008, 320, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Schwager, E.E.; Sharma, P.P.; Clarke, T.; Leite, D.J.; Wierschin, T.; Pechmann, M.; Akiyama-Oda, Y.; Esposito, L.; Bechsgaard, J.; Bilde, T.; et al. The house spider genome reveals an ancient whole-genome duplication during arachnid evolution. BMC Biol. 2017, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, W.C.; Coddington, J.A.; Crowle, L.M.; Dimitrov, D.; Goloboff, P.A.; Griswold, C.E.; Hormiga, G.; Prendini, L.; Ramirez, M.J.; Sierwald, P.; et al. The spider tree of life: Phylogeny of Araneae based on target-gene analyses from an extensive taxon sampling. Cladistics 2017, 33, 574–616. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Jurkowski, T.P.; Jeltsch, A. On the evolutionary origin of eukaryotic DNA methyltransferases and Dnmt2. PLoS ONE 2011, 6, e28104. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal-W—Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Zilberman, D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr. Biol. 2010, 20, R780–R785. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Nishimura, T. The Role of DNA methylation in transposable element silencing and genomic imprinting. In Nuclear Functions in Plant Transcription. Signaling and Development; Pontes, O., Jin, H., Eds.; Springer: New York, NY, USA, 2015; pp. 13–29. [Google Scholar]
- Xiang, H.; Zhu, J.D.; Chen, Q.; Dai, F.Y.; Li, X.; Li, M.W.; Zhang, H.Y.; Zhang, G.J.; Li, D.; Dong, Y.; et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516. [Google Scholar] [CrossRef]
- Bonasio, R.; Li, Q.Y.; Lian, J.M.; Mutti, N.S.; Jin, L.J.; Zhao, H.M.; Zhang, P.; Wen, P.; Xiang, H.; Ding, Y.; et al. Genome-wide and Caste-Specific DNA Methylomes of the Ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 2012, 22, 1755–1764. [Google Scholar] [CrossRef]
- Bewick, A.J.; Ji, L.X.; Niederhuth, C.E.; Willing, E.M.; Hofmeister, B.T.; Shi, X.L.; Wang, L.; Lu, Z.F.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef]
- Inagaki, S.; Kakutani, T. What triggers differential DNA methylation of genes and TEs: Contribution of body methylation? Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 155–160. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Kerr, A.R.W.; De Sousa, D.; Bird, A. CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 2007, 17, 625–631. [Google Scholar] [CrossRef]
- Glastad, K.M.; Gokhale, K.; Liebig, J.; Goodisman, M.A.D. The caste- and sex-specific DNA methylome of the termite Zootermopsis nevadensis. Sci. Rep. 2016, 6, 37110. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Sanchez, Z.; Mckinney, E.C.; Moore, A.J.; Moore, P.J.; Schmitz, R.J. Gene-regulatory independent functions for insect DNA methylation. Epigenetics & Chromatin 2019, 12, 6. [Google Scholar]
- Park, J.; Peng, Z.G.; Zeng, J.; Elango, N.; Park, T.; Wheeler, D.; Werren, J.H.; Yi, S.V. Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes. Mol. Biol. Evol. 2011, 28, 3345–3354. [Google Scholar] [CrossRef] [PubMed]
- Donohue, K. The epigenetics of adaptation: Focusing on epigenetic stability as an evolving trait. Evolution 2014, 68, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.I.; Spagopoulou, F. Evolutionary consequences of epigenetic inheritance. Heredity 2018, 121, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Danchin, E.; Charmantier, A.; Champagne, F.A.; Mesoudi, A.; Pujol, B.; Blanchet, S. Beyond DNA: Integrating inclusive inheritance into an extended theory of evolution. Nat. Rev. Genet. 2011, 12, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Metzger, D.C.H.; Schulte, P.M. Persistent and plastic effects of temperature on DNA methylation across the genome of threespine stickleback (Gasterosteus aculeatus). Proc. R. Soc. B-Biol. Sci. 2017, 284. [Google Scholar] [CrossRef] [PubMed]
- Metzger, D.C.H.; Schulte, P.M. The DNA methylation landscape of stickleback reveals patterns of sex chromosome evolution and effects of environmental salinity. Genome Biol. Evol. 2018, 10, 775–785. [Google Scholar] [CrossRef]
- Maor, G.L.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Gutekunst, J.; Andriantsoa, R.; Falckenhayn, C.; Hanna, K.; Stein, W.; Rasamy, J.; Lyko, F. Clonal genome evolution and rapid invasive spread of the marbled crayfish. Nat. Ecol. Evol. 2018, 2, 567–573. [Google Scholar] [CrossRef]
- Vogt, G.; Huber, M.; Thiemann, M.; van den Boogaart, G.; Schmitz, O.J.; Schubart, C.D. Production of different phenotypes from the same genotype in the same environment by developmental variation. J. Exp. Biol. 2008, 211, 510–523. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Estimated genome length | 4,287,877,091 |
| Sequence coverage | 50 |
| Assembled genome length | 2,551,871,755 |
| Number of sequences | 16,532 |
| N50 | 254,130 |
| Largest | 1,740,957 |
| GC content | 3326% |
| Number of protein-coding genes | 37601 |
| Exon length | 381 |
| Intron length | 5453 |
| Repeat content | 51,41% |
| RNA TEs | 11,4% |
| DNA TEs | 1487% |
| Unclassified | 2396% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Aagaard, A.; Bechsgaard, J.; Bilde, T. DNA Methylation Patterns in the Social Spider, Stegodyphus dumicola. Genes 2019, 10, 137. https://doi.org/10.3390/genes10020137
Liu S, Aagaard A, Bechsgaard J, Bilde T. DNA Methylation Patterns in the Social Spider, Stegodyphus dumicola. Genes. 2019; 10(2):137. https://doi.org/10.3390/genes10020137
Chicago/Turabian StyleLiu, Shenglin, Anne Aagaard, Jesper Bechsgaard, and Trine Bilde. 2019. "DNA Methylation Patterns in the Social Spider, Stegodyphus dumicola" Genes 10, no. 2: 137. https://doi.org/10.3390/genes10020137
APA StyleLiu, S., Aagaard, A., Bechsgaard, J., & Bilde, T. (2019). DNA Methylation Patterns in the Social Spider, Stegodyphus dumicola. Genes, 10(2), 137. https://doi.org/10.3390/genes10020137