Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the c. 11864G>A Nonsense Mutation

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Ophthalmological Exam
2.3. Sample Collection and DNA Extraction
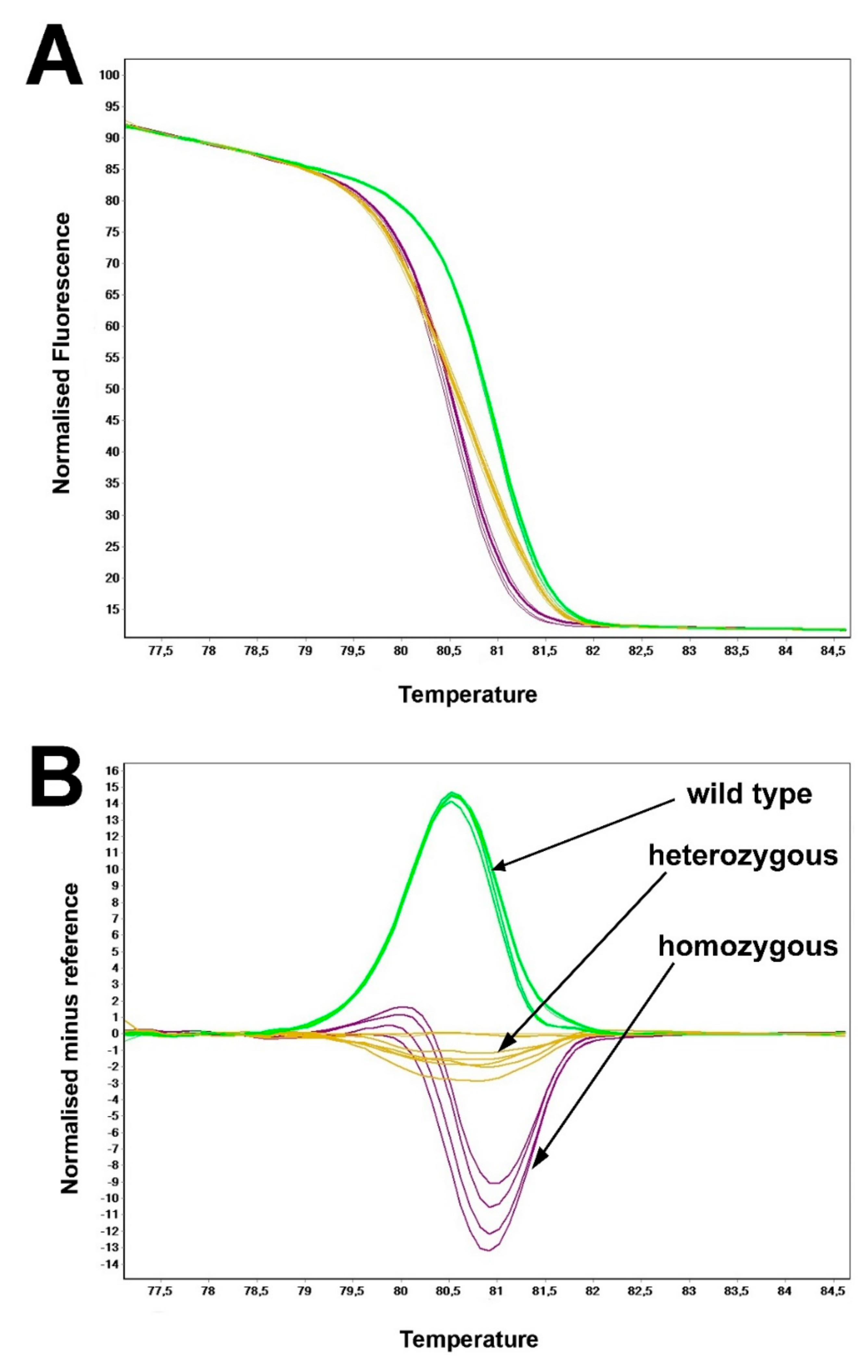
2.4. DNA Amplification and Genotyping
2.5. Haplotype Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mathur, P.; Yang, J. Usher Syndrome: Hearing Loss, Retinal Degeneration and Associated Abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed]
- López, G.; Gelvez, N.Y.; Tamayo, M. Mutational Frequencies in Usherin(Ush2a Gene) in 26 Colombian Individuals with Usher Syndrome Type Ii. Biomedica 2011, 31, 82–90. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blanco-Kelly, F.; Jaijo, T.; Aller, E.; Avila-Fernandez, A.; López-Molina, M.I.; Giménez, A.; García-Sandoval, B.; Millán, J.M.; Ayuso, C. Clinical Aspects of Usher Syndrome and the Ush2a Gene in a Cohort of 433 Patients. JAMA Ophthalmol. 2015, 133, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Bolz, H.J.; Roux, A.F. Clinical Utility Gene Card For: Usher Syndrome. Eur. J. Hum. Genet. 2011, 19, 931. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Bozbeyoglu, S.; Massof, R.W.; Kimberling, W. Natural Course of Visual Field Loss in Patients with Type 2 Usher Syndrome. Retina 2007, 27, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Fishman, G.A.; Anderson, R.J.; Grover, S.; Derlacki, D.J. Visual Acuity and Visual Field Impairment in Usher Syndrome. Arch. Ophthalmol. 1998, 116, 165–168. [Google Scholar] [CrossRef]
- Eudy, J.D.; Weston, M.D.; Yao, S.; Hoover, D.M.; Rehm, H.L.; Ma-Edmonds, M.; Yan, D.; Ahmad, I.; Cheng, J.J.; Ayuso, C.; et al. Mutation of a Gene Encoding a Protein with Extracellular Matrix Motifs in Usher Syndrome Type Iia. Science 1998, 280, 1753–1757. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin Is Required for Maintenance of Retinal Photoreceptors and Normal Development of Cochlear Hair Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef]
- Yan, D.; Ouyang, X.; Patterson, D.M.; Du, L.L.; Jacobson, S.G.; Liu, X.Z. Mutation Analysis in the Long Isoform of Ush2a in American Patients with Usher Syndrome Type Ii. J. Hum. Genet. 2009, 54, 732–738. [Google Scholar] [CrossRef]
- Weston, M.D.; Eudy, J.D.; Fujita, S.; Yao, S.; Usami, S.; Cremers, C.; Greenberg, J.; Ramesar, R.; Martini, A.; Moller, C.; et al. Genomic Structure and Identification of Novel Mutations in Usherin, the Gene Responsible for Usher Syndrome Type Iia. Am. J. Hum. Genet. 2000, 66, 1199–1210. [Google Scholar] [CrossRef]
- Van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 Novel Exons of the Usher Syndrome Type 2a (Ush2a) Gene That Encode Multiple Conserved Functional Domains and That Are Mutated in Patients with Usher Syndrome Type Ii. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.M.; Yan, D.; Hejtmancik, J.F.; Jacobson, S.G.; Li, A.R.; Du, L.L.; Angeli, S.; Kaiser, M.; Balkany, T.; Liu, X.Z. Mutational Spectrum in Usher Syndrome Type Ii. Clin. Genet. 2004, 65, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, B.; Brox, V.; Tranebjaerg, L.; Rosenberg, T.; Sadeghi, A.M.; Möller, C.; Nilssen, O. Spectrum of Ush2a Mutations in Scandinavian Patients with Usher Syndrome Type Ii. Hum. Mutat. 2008, 29, 451. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Grati, M.; Marlin, S.; Levilliers, J.; Hardelin, J.P.; Parodi, M.; Niasme-Grare, M.; Zelenika, D.; Délépine, M.; Feldmann, D.; et al. Complete Exon Sequencing of All Known Usher Syndrome Genes Greatly Improves Molecular Diagnosis. Orphanet J. Rare Dis. 2011, 6, 21. [Google Scholar] [CrossRef]
- Aller, E.; Jaijo, T.; Beneyto, M.; Nájera, C.; Oltra, S.; Ayuso, C.; Baiget, M.; Carballo, M.; Antiñolo, G.; Valverde, D.; et al. Identification of 14 Novel Mutations in the Long Isoform of Ush2a in Spanish Patients with Usher Syndrome Type Ii. J. Med. Genet. 2006, 43, e55. [Google Scholar] [CrossRef][Green Version]
- Shu, H.R.; Bi, H.; Pan, Y.C.; Xu, H.Y.; Song, J.X.; Hu, J. Targeted Exome Sequencing Reveals Novel Ush2a Mutations in Chinese Patients with Simplex Usher Syndrome. BMC Med. Genet. 2015, 16, 83. [Google Scholar] [CrossRef]
- Dreyer, B.; Tranebjaerg, L.; Rosenberg, T.; Weston, M.D.; Kimberling, W.J.; Nilssen, O. Identification of Novel Ush2a Mutations: Implications for the Structure of Ush2a Protein. Eur. J. Hum. Genet. 2000, 8, 500–506. [Google Scholar] [CrossRef]
- Dreyer, B.; Tranebjaerg, L.; Brox, V.; Rosenberg, T.; Möller, C.; Beneyto, M.; Weston, M.D.; Kimberling, W.J.; Cremers, C.W.; Liu, X.Z.; et al. A Common Ancestral Origin of the Frequent and Widespread 2299delg Ush2a Mutation. Am. J. Hum. Genet. 2001, 69, 228–234. [Google Scholar] [CrossRef]
- Baux, D.; Blanchet, C.; Hamel, C.; Meunier, I.; Larrieu, L.; Faugère, V.; Vaché, C.; Castorina, P.; Puech, B.; Bonneau, D.; et al. Enrichment of Lovd-Ushbases with 152 Ush2a Genotypes Defines an Extensive Mutational Spectrum and Highlights Missense Hotspots. Hum. Mutat. 2014, 35, 1179–1186. [Google Scholar] [CrossRef]
- Ebermann, I.; Koenekoop, R.K.; Lopez, I.; Bou-Khzam, L.; Pigeon, R.; Bolz, H.J. An Ush2a Founder Mutation Is the Major Cause of Usher Syndrome Type 2 in Canadians of French Origin and Confirms Common Roots of Quebecois and Acadians. Eur. J. Hum. Genet. 2009, 17, 80–84. [Google Scholar] [CrossRef]
- Aller, E.; Larrieu, L.; Jaijo, T.; Baux, D.; Espinós, C.; González-Candelas, F.; Nájera, C.; Palau, F.; Claustres, M.; Roux, A.F.; et al. The Ush2a C.2299delg Mutation: Dating Its Common Origin in a Southern European Population. Eur. J. Hum. Genet. 2010, 18, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefèvre, G.M.; Hardelin, J.P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An Innovative Strategy for the Molecular Diagnosis of Usher Syndrome Identifies Causal Biallelic Mutations in 93% of European Patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Bolz, H.; Neuhaus, C.; Preising, M.; Khan, A.; Gliem, M.; Charbel Issa, P.; Wolfrum, U.; Gal, A.; Lorenz, B.; Eisenberger, T. High-Coverage Next-Generation Sequencing (Ngs) for Retinal Dystrophies and Usher Syndrome: High Diagnostic Yield, Cnv Detection, Novel Disease Mechanisms and Therapy Targets. Invest. Ophthalmol. Vis. Sci. 2013, 54, 3373. [Google Scholar]
- Aller, E.; Nájera, C.; Millán, J.M.; Oltra, J.S.; Pérez-Garrigues, H.; Vilela, C.; Navea, A.; Beneyto, M. Genetic Analysis of 2299delg and C759f Mutations (Ush2a) in Patients with Visual and/or Auditory Impairments. Eur. J. Hum. Genet. 2004, 12, 407–410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V.; Windsor, E.A.; Sumaroka, A.; Roman, A.J.; Rane, T.; Smilko, E.E.; Bennett, J.; Stone, E.M.; et al. Disease Expression in Usher Syndrome Caused by Vlgr1 Gene Mutation (Ush2c) and Comparison with Ush2a Phenotype. Invest. Ophthalmol. Vis. Sci. 2005, 46, 734–743. [Google Scholar] [CrossRef]
- Bernal, S.; Medà, C.; Solans, T.; Ayuso, C.; Garcia-Sandoval, B.; Valverde, D.; Del Rio, E.; Baiget, M. Clinical and Genetic Studies in Spanish Patients with Usher Syndrome Type Ii: Description of New Mutations and Evidence for a Lack of Genotype--Phenotype Correlation. Clin. Genet. 2005, 68, 204–214. [Google Scholar] [CrossRef]
- Ebermann, I.; Phillips, J.B.; Liebau, M.C.; Koenekoop, R.K.; Schermer, B.; Lopez, I.; Schäfer, E.; Roux, A.-F.; Dafinger, C.; Bernd, A.; et al. Pdzd7 Is a Modifier of Retinal Disease and a Contributor to Digenic Usher Syndrome. J. Clin. Investig. 2010, 120, 1812–1823. [Google Scholar] [CrossRef]
- Abadie, C.; Blanchet, C.; Baux, D.; Larrieu, L.; Besnard, T.; Ravel, P.; Biboulet, R.; Hamel, C.; Malcolm, S.; Mondain, M.; et al. Audiological Findings in 100 Ush2 Patients. Clin. Genet. 2012, 82, 433–438. [Google Scholar] [CrossRef]
- Vossen, R.H. Genotyping DNA Variants with High-Resolution Melting Analysis. Methods Mol. Biol. 2017, 1492, 17–28. [Google Scholar]
- Fakin, A.; Jarc-Vidmar, M.; Glavac, D.; Bonnet, C.; Petit, C.; Hawlina, M. Fundus Autofluorescence and Optical Coherence Tomography in Relation to Visual Function in Usher Syndrome Type 1 and 2. Vision Res. 2012, 75, 60–70. [Google Scholar] [CrossRef]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; McGee, T.L.; Dryja, T.P.; Berson, E.L. Disease Course in Patients with Autosomal Recessive Retinitis Pigmentosa Due to the Ush2a Gene. Invest. Ophthalmol. Vis. Sci. 2008, 49, 5532–5539. [Google Scholar] [CrossRef]
- Testa, F.; Melillo, P.; Bonnet, C.; Marcelli, V.; de Benedictis, A.; Colucci, R.; Gallo, B.; Kurtenbach, A.; Rossi, S.; Marciano, E.; et al. Clinical Presentation and Disease Course of Usher Syndrome Because of Mutations in Myo7a or Ush2a. Retina 2017, 37, 1581–1590. [Google Scholar] [CrossRef]
- Hartel, B.P.; Lofgren, M.; Huygen, P.L.; Guchelaar, I.; Lo-A-Njoe Kort, N.; Sadeghi, A.M.; van Wijk, E.; Tranebjaerg, L.; Kremer, H.; Kimberling, W.J.; et al. A Combination of Two Truncating Mutations in Ush2a Causes More Severe and Progressive Hearing Impairment in Usher Syndrome Type Iia. Hear. Res. 2016, 339, 60–68. [Google Scholar] [CrossRef]
- Pierrache, L.H.; Hartel, B.P.; van Wijk, E.; Meester-Smoor, M.A.; Cremers, F.P.; de Baere, E.; de Zaeytijd, J.; van Schooneveld, M.J.; Cremers, C.W.; Dagnelie, G.; et al. Visual Prognosis in Ush2a-Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type Iia Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2016, 123, 1151–1160. [Google Scholar] [CrossRef]
- Nagase, Y.; Kurata, K.; Hosono, K.; Suto, K.; Hikoya, A.; Nakanishi, H.; Mizuta, K.; Mineta, H.; Minoshima, S.; Hotta, Y. Visual Outcomes in Japanese Patients with Retinitis Pigmentosa and Usher Syndrome Caused by Ush2a Mutations. Semin. Ophthalmol. 2018, 33, 560–565. [Google Scholar] [CrossRef]
- Zhang, Z.; Xin, D.; Wang, P.; Zhou, L.; Hu, L.; Kong, X.; Hurst, L.D. Noisy Splicing, More Than Expression Regulation, Explains Why Some Exons Are Subject to Nonsense-Mediated Mrna Decay. BMC Biol. 2009, 7, 23. [Google Scholar] [CrossRef]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-Based Next Generation Sequencing as a Reliable and Efficient Technique to Detect Mutations in Unselected Patients with Retinal Dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef]
- Ivanova, M.E.; Trubilin, V.N.; Atarshchikov, D.S.; Demchinsky, A.M.; Strelnikov, V.V.; Tanas, A.S.; Orlova, O.M.; Machalov, A.S.; Overchenko, K.V.; Markova, T.V.; et al. Genetic screening of Russian Usher syndrome patients toward selection for gene therapy. Ophthalmic Genet. 2018, 39, 706–713. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Exon | Primer Sequence | Final Concentration (nM) | |
|---|---|---|---|
| 61 | Forward: | 5’-TCTGGTCAGAAGGAGCCCTTGAAT-3’ | 200 |
| Reverse: | 5’-AGGTGGAGCTTCCAGAGTTTGTGT-3’ | ||
| 11–14 | 11–32 | 11–33 | 11–44 | 12–36 | 12–37 | 12–56 | 12–79 | 12–88 | 02–06 | |
|---|---|---|---|---|---|---|---|---|---|---|
| rs10779261 | C | T | T | T | T | T | T | C/T | T | C/T |
| rs4253963 | T | C | C | C | C/T | C | C | C/T | C | C/T |
| rs1805050 | G | G | G | G | G | G | G | G | G | A/G |
| rs1324330 | T | T | T | T | C/T | T | T | T | T | C/T |
| rs646094 | A | A | A | A | A | A | A | A | A | A/C |
| rs1805049 | T | T | T | T | C/T | T | T | T | T | C/T |
| rs6657250 | G | G | G | G | G | G | G | G | G | G |
| rs10864219 | A | A | A | A | A/G | A | A | A | A | A |
| rs10864198 | T | T | T | T | T | T | T | T | T | T |
| rs11120616 | G | G | G | G | G | G | G | G | G | G |
| rs35309576 | T | T | T | T | T | T | T | T | T | T |
| rs2820718 | C | C | C | C | C | C | C | C | C | C |
| Haplotype | Ht1 | Ht2 | Ht2 | Ht2 | Ht2/3 | Ht2 | Ht2 | Ht1/2 | Ht2 | Ht1/4 |
| Patient | Sex | Age at Onset of Nyctalopia (Years) | Age at Eye Exam (Years) | VA (RE, LE) | Ishihara (RE, LE; N Out of 15 Plates Seen) | Goldmann II4 Radius (RE, LE) (Degrees) | Fundus Autofluorescence Pattern (RE, LE) | Age at Audiogram (Years) | Average Hearing Loss Accross (R,L) (dB) |
|---|---|---|---|---|---|---|---|---|---|
| 2–06 | Male | 12 | 19 | 1.0. 1.0 | N/A | N/A | Ring, Ring | 21 | 56, 60 |
| 12–79 | Male | 22 | 28 | 1.0, 1.0 | 14, 14 | 10, 15 | Ring, Ring | 17 | 66, 68 |
| 11–33 | Male | 9 | 38 | 0.8, 0.9 | 13, 13 | 11, 10 | Ring, Ring | 30 | 130*, 80 |
| 11–44 | Male | 13 | 41 | 0.6, 0.6 | 1, 1 | 3, 3 | Ring, Ring | 42 | 71, 65 |
| 11–14 | Female | 12 | 44 | 0.15, 0.2 | 0, 0 | 0, 0 | Patch, Patch | 44 | 59, 68 |
| 11–32 | Male | 10 | 61 | CF, HM | 0, 0 | 2, 1 | Atrophy, Atrophy | 57 | 75, 75 |
| 12–56 | Male | 42 | 62 | 0.6, 0.2 | 3, 1 | 6, 6 | Ring, Ring | N/A | N/A |
| 12–37 | Male | 16 | 64 | LP, LP | 0, 0 | 0, 0 | Patch, Patch | 65 | 59, 61 |
| 12–36 | Female | 35 | 65 | HM, HM | 0, 0 | 0, 0 | Patch, Patch | N/A | N/A |
| 12–-8 | Female | 4 | 73 | HM, HM | 0, 0 | N/A | Patch, Atrophy | 63 | 89, 98 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zupan, A.; Fakin, A.; Battelino, S.; Jarc-Vidmar, M.; Hawlina, M.; Bonnet, C.; Petit, C.; Glavač, D. Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the c. 11864G>A Nonsense Mutation. Genes 2019, 10, 1015. https://doi.org/10.3390/genes10121015
Zupan A, Fakin A, Battelino S, Jarc-Vidmar M, Hawlina M, Bonnet C, Petit C, Glavač D. Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the c. 11864G>A Nonsense Mutation. Genes. 2019; 10(12):1015. https://doi.org/10.3390/genes10121015
Chicago/Turabian StyleZupan, Andrej, Ana Fakin, Saba Battelino, Martina Jarc-Vidmar, Marko Hawlina, Crystel Bonnet, Christine Petit, and Damjan Glavač. 2019. "Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the c. 11864G>A Nonsense Mutation" Genes 10, no. 12: 1015. https://doi.org/10.3390/genes10121015
APA StyleZupan, A., Fakin, A., Battelino, S., Jarc-Vidmar, M., Hawlina, M., Bonnet, C., Petit, C., & Glavač, D. (2019). Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the c. 11864G>A Nonsense Mutation. Genes, 10(12), 1015. https://doi.org/10.3390/genes10121015