Transcriptomic Analysis Reveals Involvement of the Macrophage Migration Inhibitory Factor Gene Network in Duchenne Muscular Dystrophy

 , , ,
, , ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Network Construction
2.2. Dataset Selection and Analysis
2.3. Statistical Analysis
3. Results
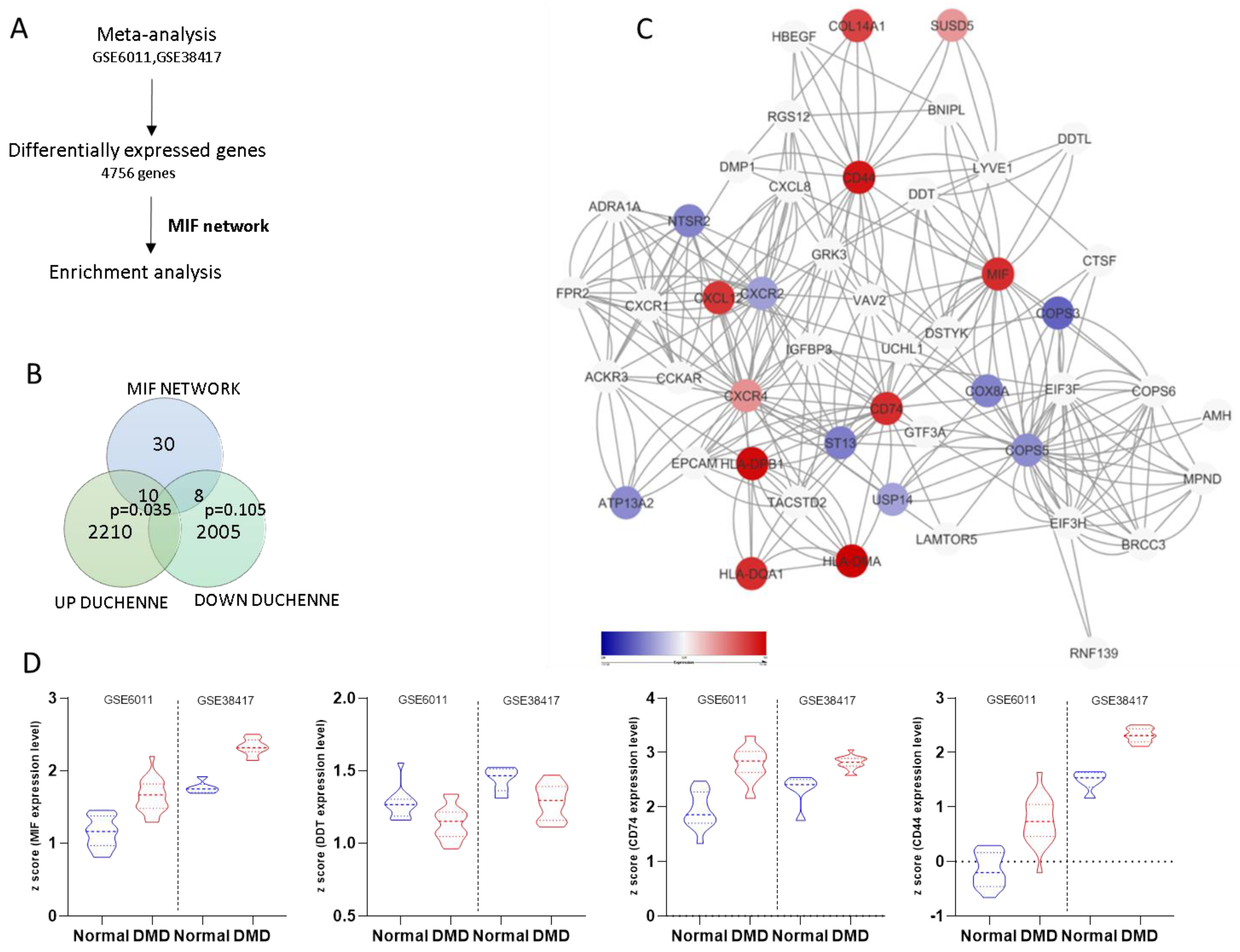
3.1. Generation of the MIF Network
3.2. Meta-Analysis of Gene Expression in Duchenne Muscular Dystrophy
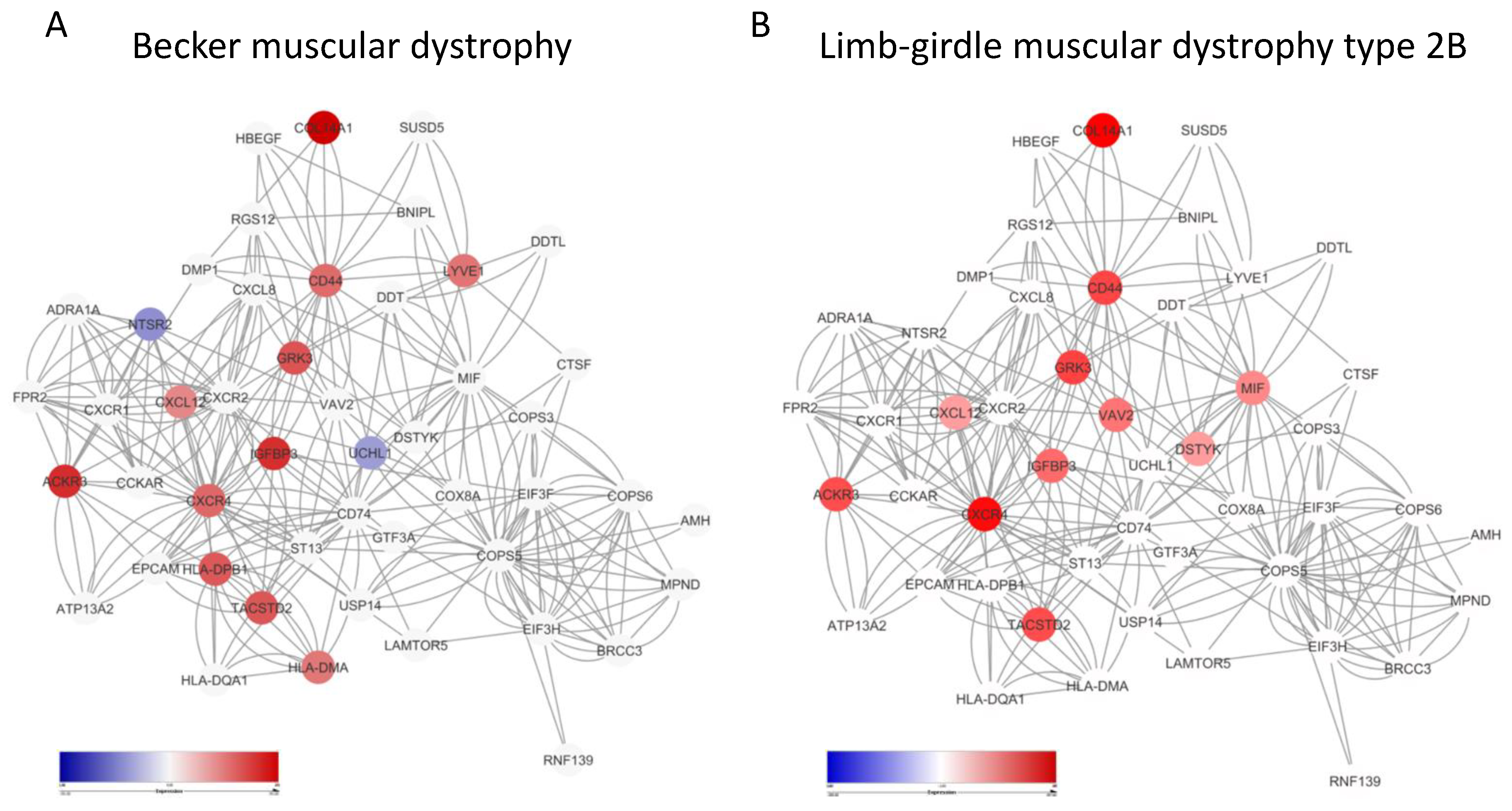
3.3. Enrichment of the MIF Network in Becker Disease and Limb-Girdle Muscular Dystrophy Type 2B
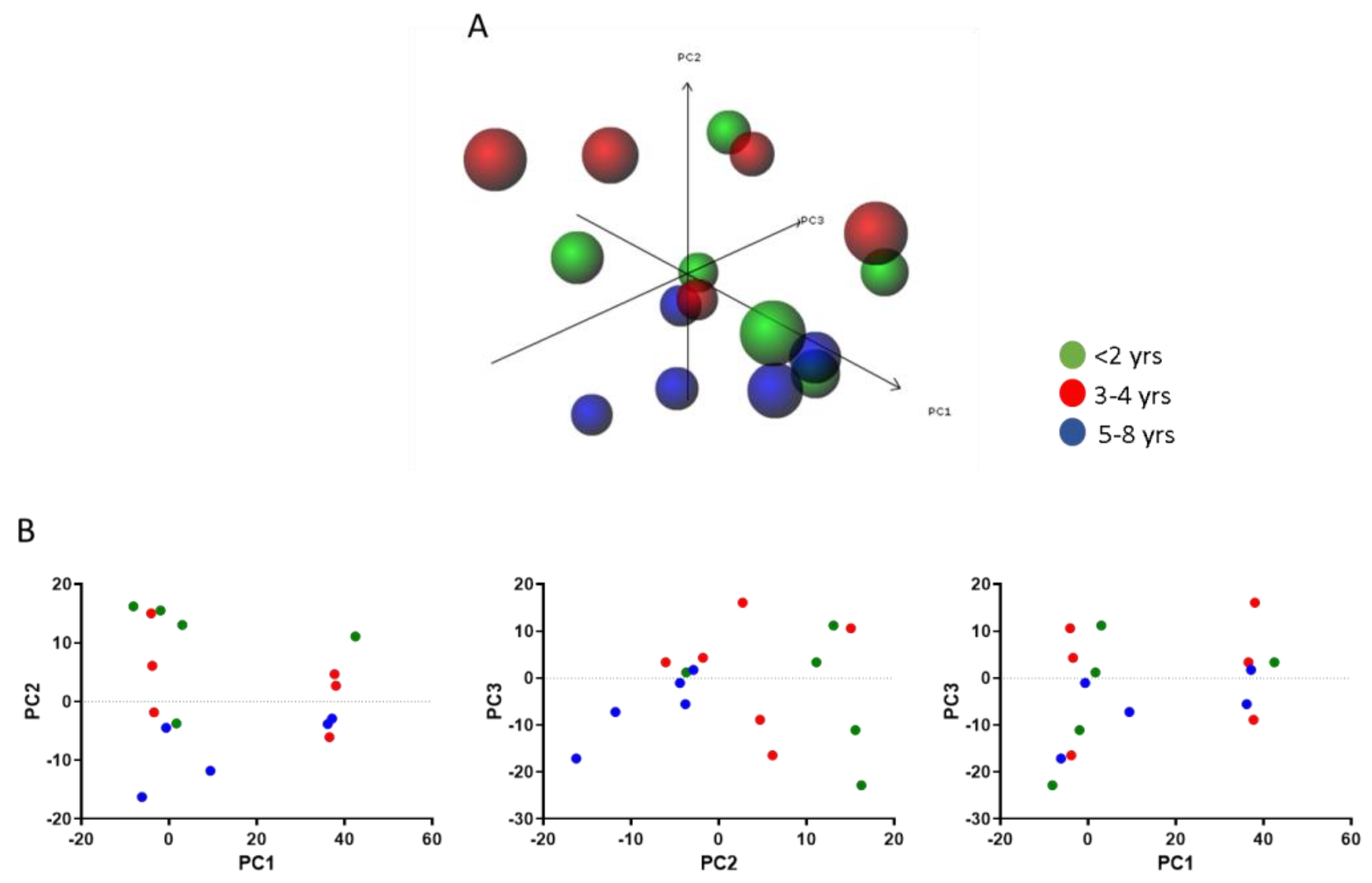
3.4. Modulation of the MIF Pathway in Muscle Biopsies of DMD Patients at Different Ages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health 2015, 51, 759–764. [Google Scholar] [CrossRef]
- Liang, Y.; Chen, S.; Zhu, J.; Zhou, X.; Yang, C.; Yao, L.; Zhang, C. Dystrophin hydrophobic regions in the pathogenesis of duchenne and becker muscular dystrophies. Bosn. J. Basic Med. Sci. 2015, 15, 42. [Google Scholar] [CrossRef]
- Le Rumeur, E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn. J. Basic Med. Sci. 2015, 15, 14–20. [Google Scholar] [CrossRef]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-inflammatory drugs for duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 2016, 10, 2745. [Google Scholar]
- Sreetama, S.C.; Chandra, G.; Van der Meulen, J.H.; Ahmad, M.M.; Suzuki, P.; Bhuvanendran, S.; Nagaraju, K.; Hoffman, E.P.; Jaiswal, J.K. Membrane Stabilization by Modified Steroid Offers a Potential Therapy for Muscular Dystrophy Due to Dysferlin Deficit. Mol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidbal, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Mojumdar, K.; Liang, F.; Lemaire, C.; Li, T.; Richardson, J.; Divangahi, M.; Qureshi, S.; Petrof, B.J. Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum. Mol. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR 2. EMBO Mol. Med. 2014. [Google Scholar] [CrossRef]
- Fagone, P.; Mazzon, E.; Cavalli, E.; Bramanti, A.; Petralia, M.C.; Mangano, K.; Al-Abed, Y.; Bramati, P.; Nicoletti, F. Contribution of the macrophage migration inhibitory factor superfamily of cytokines in the pathogenesis of preclinical and human multiple sclerosis: In silico and in vivo evidences. J. Neuroimmunol. 2018, 322. [Google Scholar] [CrossRef]
- Nicoletti, F.; Créange, A.; Orlikowski, D.; Bolgert, F.; Mangano, K.; Metz, C.; Di Marco, R.; Al Abed, Y. Macrophage migration inhibitory factor (MIF) seems crucially involved in Guillain-Barré syndrome and experimental allergic neuritis. J. Neuroimmunol. 2005, 168, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mangano, K.; Di Marco, R.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Upregulated Expression of Macrophage Migration Inhibitory Factor, Its Analogue D-Dopachrome Tautomerase, and the CD44 Receptor in Peripheral CD4 T Cells from Clinically Isolated Syndrome Patients with Rapid Conversion to Clinical Defined Multiple Sclerosis. Medicina 2019, 55, 667. [Google Scholar] [CrossRef] [PubMed]
- Presti, M.; Mazzon, E.; Basile, M.S.; Petralia, M.C.; Bramanti, A.; Colletti, G.; Bramanti, P.; Nicoletti, F.; Fagone, P. Overexpression of macrophage migration inhibitory factor and functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in glioblastoma. Oncol. Lett. 2018, 16, 2881–2886. [Google Scholar] [CrossRef] [PubMed]
- Mangano, K.; Mazzon, E.; Basile, M.S.; Di Marco, R.; Bramanti, P.; Mammana, S.; Petralia, M.C.; Fagone, P.; Nicoletti, F. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 2018, 9, 17951–17970. [Google Scholar] [CrossRef]
- Günther, S.; Fagone, P.; Jalce, G.; Atanasov, A.G.; Guignabert, C.; Nicoletti, F. Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: From pathogenic factors to therapeutic targets. Drug Discov. Today 2019, 24, 428–439. [Google Scholar] [CrossRef]
- Reimann, J.; Schnell, S.; Schwartz, S.; Kappes-Horn, K.; Dodel, R.; Bacher, M. Macrophage migration inhibitory factor in normal human skeletal muscle and inflammatory myopathies. J. Neuropathol. Exp. Neurol. 2010. [Google Scholar] [CrossRef]
- Miyatake, S.; Manabe, Y.; Inagaki, A.; Furuichi, Y.; Takagi, M.; Taoka, M.; Isobe, T.; Hirota, K.; Fujii, N.L. Macrophage migration inhibitory factor diminishes muscle glucose transport induced by insulin and AICAR in a muscle type-dependent manner. Biochem. Biophys. Res. Commun. 2014. [Google Scholar] [CrossRef]
- Subbannayya, T.; Variar, P.; Advani, J.; Nair, B.; Shankar, S.; Gowda, H.; Saussez, S.; Chatterjee, A.; Prasad, T.S.K. An integrated signal transduction network of macrophage migration inhibitory factor. J. Cell Commun. Signal. 2016. [Google Scholar] [CrossRef]
- Zuberi, K.; Franz, M.; Rodriguez, H.; Montojo, J.; Lopes, C.T.; Bader, G.D.; Morris, Q. GeneMANIA Prediction Server 2013 Update. Nucleic Acids Res. 2013, 41, W115–W122. [Google Scholar] [CrossRef]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef]
- Toro-Domínguez, D.; Martorell-Marugán, J.; López-Domínguez, R.; García-Moreno, A.; González-Rumayor, V.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. ImaGEO: Integrative gene expression meta-analysis from GEO database. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.-W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Thorley, M.; Duguez, S.; Mazza, E.M.C.; Valsoni, S.; Bigot, A.; Mamchaoui, K.; Harmon, B.; Voit, T.; Mouly, V.; Duddy, W. Skeletal muscle characteristics are preserved in hTERT/cdk4 human myogenic cell lines. Skelet. Muscle 2016, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Gutiérrez-Pabello, J.A.; Kramnik, I.; Maiti, T.; Quackenbush, J. An improved empirical bayes approach to estimating differential gene expression in microarray time-course data: BETR (Bayesian Estimation of Temporal Regulation). BMC Bioinform. 2009, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A Free, Open-Source System for Microarray Data Management and Analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Mazzon, E.; Basile, M.S.; Campo, G.; Corsico, F.; Presti, M.; Bramanti, P.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; et al. Modulation of Tetraspanin 32 (TSPAN32) Expression in T Cell-Mediated Immune Responses and in Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 4323. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Bramanti, P.; Nania, R.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Upregulation of IL-1 Receptor Antagonist in a Mouse Model of Migraine. Brain Sci. 2019, 9, 172. [Google Scholar] [CrossRef]
- Fagone, P.; Mazzon, E.; Mammana, S.; Di Marco, R.; Spinasanta, F.; Basile, M.; Petralia, M.; Bramanti, P.; Nicoletti, F.; Mangano, K. Identification of CD4+ T cell biomarkers for predicting the response of patients with relapsing‑remitting multiple sclerosis to natalizumab treatment. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Falzone, L.; Bramanti, P.; Nicoletti, F.; Basile, M.S. Retrospective follow-up analysis of the transcriptomic patterns of cytokines, cytokine receptors and chemokines at preconception and during pregnancy, in women with post-partum depression. Exp. Ther. Med. 2019, 18, 2055–2062. [Google Scholar] [CrossRef]
- Nicoletti, F.; Mazzon, E.; Fagone, P.; Mangano, K.; Mammana, S.; Cavalli, E.; Basile, M.S.; Bramanti, P.; Scalabrino, G.; Lange, A.; et al. Prevention of clinical and histological signs of MOG-induced experimental allergic encephalomyelitis by prolonged treatment with recombinant human EGF. J. Neuroimmunol. 2019, 332, 224–232. [Google Scholar] [CrossRef]
- Candido, S.; Lupo, G.; Pennisi, M.; Basile, M.; Anfuso, C.; Petralia, M.; Gattuso, G.; Vivarelli, S.; Spandidos, D.; Libra, M.; et al. The analysis of miRNA expression profiling datasets reveals inverse microRNA patterns in glioblastoma and Alzheimer’s disease. Oncol. Rep. 2019, 42, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.D.; Presti, M.; Mangano, K.; Petralia, M.C.; Basile, M.S.; Libra, M.; Candido, S.; Fagone, P.; Mazzon, E.; Nicoletti, F.; et al. Prediction of PD-L1 Expression in Neuroblastoma via Computational Modeling. Brain Sci. 2019, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Basile, M.S.; Mazzon, E.; Russo, A.; Mammana, S.; Longo, A.; Bonfiglio, V.; Fallico, M.; Caltabiano, R.; Fagone, P.; Nicoletti, F.; et al. Differential modulation and prognostic values of immune-escape genes in uveal melanoma. PLoS ONE 2019, 14, e0210276. [Google Scholar] [CrossRef] [PubMed]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Russo, A.; Longo, A.; Avitabile, T.; Nicoletti, F.; Reibaldi, M.; Basile, M.S. Characterization of the Pathophysiological Role of CD47 in Uveal Melanoma. Molecules 2019, 24, 2450. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Caltabiano, R.; Russo, A.; Lupo, G.; Anfuso, C.D.; Basile, M.S.; Longo, A.; Nicoletti, F.; De Pasquale, R.; Libra, M.; et al. Identification of novel chemotherapeutic strategies for metastatic uveal melanoma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Mammana, S.; Bramanti, P.; Mazzon, E.; Cavalli, E.; Basile, M.S.; Fagone, P.; Petralia, M.C.; McCubrey, J.A.; Nicoletti, F.; Mangano, K. Preclinical evaluation of the PI3K/Akt/mTOR pathway in animal models of multiple sclerosis. Oncotarget 2018, 9, 8263–8277. [Google Scholar] [CrossRef]
- Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.S.; Petralia, M.C.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The role of macrophages in neuroinflammatory and neurodegenerative pathways of alzheimer’s disease, amyotrophic lateral sclerosis, and multiple sclerosis: Pathogenetic cellular effectors and potential therapeutic targets. Int. J. Mol. Sci. 2018, 19, 831. [Google Scholar] [CrossRef]
- Parolo, S.; Marchetti, L.; Lauria, M.; Misselbeck, K.; Scott-Boyer, M.P.; Caberlotto, L.; Priami, C. Combined use of protein biomarkers and network analysis unveils deregulated regulatory circuits in Duchenne muscular dystrophy. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33. [Google Scholar] [CrossRef]
- Yoo, S.A.; Leng, L.; Kim, B.J.; Du, X.; Tilstam, P.V.; Kim, K.H.; Kong, J.S.; Yoon, H.J.; Liu, A.; Wang, T.; et al. MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef]
- Morand, E.F.; Leech, M.; Bernhagen, J. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 2006, 5, 399. [Google Scholar] [CrossRef] [PubMed]
- Gore, Y.; Starlets, D.; Maharshak, N.; Becker-Herman, S.; Kaneyuki, U.; Leng, L.; Bucala, R.; Shachar, I. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J. Biol. Chem. 2008. [Google Scholar] [CrossRef] [PubMed]
- Schneppenheim, J.; Dressel, R.; Hüttl, S.; Lüllmann-Rauch, R.; Engelke, M.; Dittmann, K.; Wienands, J.; Eskelinen, E.L.; Hermans-Borgmeyer, I.; Fluhrer, R.; et al. The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J. Exp. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lantner, F.; Starlets, D.; Gore, Y.; Flaishon, L.; Yamit-Hezi, A.; Dikstein, R.; Leng, L.; Bucala, R.; Machluf, Y.; Oren, M.; et al. CD74 induces TAp63 expression leading to B-cell survival. Blood 2007. [Google Scholar] [CrossRef] [PubMed]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Müller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, H. Targeting fibrosis in duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in duchenne muscular dystrophy. Biomed Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Ogura, Y.; Tajrishi, M.M.; Sato, S.; Hindi, S.M.; Kumar, A. Therapeutic potential of matrix metalloproteinases in Duchenne muscular dystrophy. Front. Cell Dev. Biol. 2014, 2, 11. [Google Scholar] [CrossRef]
- Günther, S.; Bordenave, J.; Hua-Huy, T.; Nicco, C.; Cumont, A.; Thuillet, R.; Tu, L.; Quatremarre, T.; Guilbert, T.; Jalce, G.; et al. Macrophage Migration Inhibitory Factor (MIF) Inhibition in a Murine Model of Bleomycin-Induced Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 4105. [Google Scholar] [CrossRef]
- Escolar, D.M.; Hache, L.P.; Clemens, P.R.; Cnaan, A.; McDonald, C.M.; Viswanathan, V.; Kornberg, A.J.; Bertorini, T.E.; Nevo, Y.; Lotze, T.; et al. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology 2011. [Google Scholar] [CrossRef]
- Serra, F.; Quarta, M.; Canato, M.; Toniolo, L.; De Arcangelis, V.; Trotta, A.; Spath, L.; Monaco, L.; Reggiani, C.; Naro, F. Inflammation in muscular dystrophy and the beneficial effects of non-steroidal anti-inflammatory drugs. Muscle Nerve 2012, 46, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.L.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-κB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Investig. 2007. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Crichlow, G.V.; Vermeire, J.J.; Leng, L.; Du, X.; Hodsdon, M.E.; Bucala, R.; Cappello, M.; Gross, M.; Gaeta, F.; et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc. Natl. Acad. Sci. USA 2010, 107, 11313–11318. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef]
- Haran, M.; Mirkin, V.; Braester, A.; Harpaz, N.; Shevetz, O.; Shtreiter, M.; Greenberg, S.; Mordich, O.; Amram, O.; Binsky-Ehrenreich, I.; et al. A phase I-II clinical trial of the anti-CD74 monoclonal antibody milatuzumab in frail patients with refractory chronic lymphocytic leukaemia: A patient based approach. Br. J. Haematol. 2018, 182, 125–128. [Google Scholar] [CrossRef]
- Nakahara, K.; Fujikawa, K.; Hiraoka, H.; Miyazaki, I.; Asanuma, M.; Ito, A.; Takasugi, N.; Uehara, T. Attenuation of Macrophage Migration Inhibitory Factor-Stimulated Signaling via S-Nitrosylation. Biol. Pharm. Bull. 2019, 42, 1044–1047. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Therapeutic strategies to address neuronal nitric oxide synthase deficiency and the loss of nitric oxide bioavailability in Duchenne Muscular Dystrophy. Orphanet J. Rare Dis. 2017, 12, 100. [Google Scholar] [CrossRef]
- Thomas, G.D.; Ye, J.; De Nardi, C.; Monopoli, A.; Ongini, E.; Victor, R.G. Treatment with a nitric oxide-donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS ONE 2012, 7, e49350. [Google Scholar] [CrossRef]
- Paskas, S.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Al-Abed, Y.; He, M.; Rakocevic, S.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Lopinavir-NO, a nitric oxide-releasing HIV protease inhibitor, suppresses the growth of melanoma cells in vitro and in vivo. Investig. New Drugs 2019, 37, 1014–1028. [Google Scholar] [CrossRef]
- Basile, M.; Mazzon, E.; Krajnovic, T.; Draca, D.; Cavalli, E.; Al-Abed, Y.; Bramanti, P.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Anticancer and Differentiation Properties of the Nitric Oxide Derivative of Lopinavir in Human Glioblastoma Cells. Molecules 2018, 23, 2463. [Google Scholar] [CrossRef]
- Maksimovic-Ivanic, D.; Mojic, M.; Bulatovic, M.; Radojkovic, M.; Kuzmanovic, M.; Ristic, S.; Stosic-Grujicic, S.; Miljkovic, D.; Cavalli, E.; Libra, M.; et al. The NO-modified HIV protease inhibitor as a valuable drug for hematological malignancies: Role of p70S6K. Leuk. Res. 2015, 39, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Paskaš, S.; Krajnović, T.; Basile, M.S.; Dunđerović, D.; Cavalli, E.; Mangano, K.; Mammana, S.; Al-Abed, Y.; Nicoletti, F.; Mijatović, S.; et al. Senescence as a main mechanism of Ritonavir and Ritonavir-NO action against melanoma. Mol. Carcinog. 2019, 58, 1362–1375. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Cavalli, E.; Mammana, S.; Lombardo, G.A.G.; Pennisi, V.; Zocca, M.-B.; He, M.; Al-Abed, Y.; et al. Effects of NO-Hybridization on the Immunomodulatory Properties of the HIV Protease Inhibitors Lopinavir and Ritonavir. Basic Clin. Pharmacol. Toxicol. 2015, 117, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Takagi, H.; Horiguchi, N.; Toyoda, M.; Kanda, D.; Otsuka, T.; Emoto, Y.; Emoto, M.; Mori, M. Lack of macrophage migration inhibitory factor protects mice against concanavalin A-induced liver injury. Liver Int. 2006, 26, 346–351. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Symbol | p-Values | Adj-p-Values | Log Fold-Change | t-Statistic |
|---|---|---|---|---|
| Ackr3 | 0.7736 | 0.9997 | −1.0993 | −0.2921 |
| Adra1a | 0.9940 | 0.9997 | 0.0286 | 0.0076 |
| Amh | 0.9782 | 0.9997 | 0.1045 | 0.0277 |
| Atp13a2 | 0.9363 | 0.9997 | −0.3327 | −0.0810 |
| Bnipl | 0.9948 | 0.9997 | 0.0252 | 0.0066 |
| Brcc3 | 0.9778 | 0.9997 | −0.1144 | −0.0282 |
| Cckar | 0.9988 | 0.9997 | −0.0058 | −0.0015 |
| Cd44 | 0.9900 | 0.9997 | −0.0764 | −0.0127 |
| Cd74 | 0.9584 | 0.9997 | 0.1988 | 0.0529 |
| Col14a1 | 0.9821 | 0.9997 | 0.0863 | 0.0227 |
| Cops3 | 0.9938 | 0.9997 | −0.0417 | −0.0079 |
| Cops5 | 0.9967 | 0.9997 | −0.0224 | −0.0042 |
| Cops6 | 0.9788 | 0.9997 | −0.1265 | −0.0269 |
| Cox8a | 0.9928 | 0.9997 | 0.0541 | 0.0092 |
| Ctsf | 0.8714 | 0.9997 | 0.6937 | 0.1642 |
| Cxcl12 | 0.8103 | 0.9997 | 1.1162 | 0.2436 |
| Cxcl8 | 0.7477 | 0.9997 | 1.6641 | 0.3266 |
| Cxcr1 | 0.9895 | 0.9997 | 0.0503 | 0.0133 |
| Cxcr2 | 0.9931 | 0.9997 | −0.0332 | −0.0088 |
| Cxcr4 | 0.9865 | 0.9997 | 0.0641 | 0.0171 |
| Ddt | 0.9830 | 0.9997 | −0.1208 | −0.0216 |
| Ddtl | 0.9864 | 0.9997 | −0.0649 | −0.0173 |
| Dmp1 | 0.9995 | 0.9997 | −0.0025 | −0.0007 |
| Dstyk | 0.9135 | 0.9997 | −0.4908 | −0.1102 |
| Dusp14 | 0.9551 | 0.9997 | 0.2850 | 0.0571 |
| Eif3f | 0.9699 | 0.9997 | −0.2166 | −0.0382 |
| Eif3h | 0.9997 | 0.9997 | 0.0023 | 0.0004 |
| Epcam | 0.9901 | 0.9997 | 0.0475 | 0.0125 |
| Fpr2 | 0.9989 | 0.9997 | 0.0050 | 0.0013 |
| Hla–Dma | 0.9984 | 0.9997 | −0.0082 | −0.0020 |
| Hla–Dpb1 | 0.9938 | 0.9997 | 0.0293 | 0.0078 |
| Hla–Dqa1 | 0.9960 | 0.9997 | 0.0190 | 0.0051 |
| Igfbp3 | 0.8091 | 0.9997 | 1.3739 | 0.2452 |
| Lamtor5 | 0.9795 | 0.9997 | −0.1449 | −0.0261 |
| Lyve1 | 0.9951 | 0.9997 | 0.0232 | 0.0062 |
| Mif | 0.9855 | 0.9997 | −0.1113 | −0.0184 |
| Mpnd | 0.9990 | 0.9997 | 0.0048 | 0.0013 |
| Ntsr2 | 0.9979 | 0.9997 | 0.0102 | 0.0027 |
| Rgs12 | 0.9713 | 0.9997 | 0.1623 | 0.0365 |
| Rnf139 | 0.9994 | 0.9997 | 0.0029 | 0.0008 |
| St13 | 0.9808 | 0.9997 | 0.1248 | 0.0244 |
| Susd5 | 0.9952 | 0.9997 | −0.0235 | −0.0061 |
| Tacstd2 | 0.8993 | 0.9997 | 0.4954 | 0.1284 |
| Uchl1 | 0.9456 | 0.9997 | −0.3802 | −0.0692 |
| Vav2 | 0.9707 | 0.9997 | 0.1509 | 0.0373 |
| Gene Symbol | ID_REF | Significance-Values |
|---|---|---|
| Ackr3 | 1559114_a_at | 0.999979 |
| Adra1a | 211489_at | 0.999979 |
| Amh | 206516_at | 0.999981 |
| Atp13a2 | 218608_at | 0.99998 |
| Brcc3 | 231913_s_at | 0.99998 |
| Cckar | 211174_s_at | 0.99998 |
| Cd44 | 212063_at | 0.999968 |
| Cd74 | 1567627_at | 0.999978 |
| Col14a1 | 212865_s_at | 0.927757 |
| Cops3 | 202078_at | 0.999981 |
| Cops5 | 201652_at | 0.999974 |
| Cops6 | 201405_s_at | 0.99998 |
| Cox8a | 201119_s_at | 0.99998 |
| Ctsf | 203657_s_at | 0.99998 |
| Cxcl12 | 209687_at | 0.999979 |
| Cxcl8 | 202859_x_at | 0.99998 |
| Cxcr1 | 207094_at | 0.999979 |
| Cxcr2 | 207008_at | 0.999976 |
| Cxcr4 | 217028_at | 0.999979 |
| Ddt | 202929_s_at | 0.99998 |
| Dmp1 | 217067_s_at | 0.999981 |
| Dstyk | 211515_s_at | 0.999979 |
| Eif3f | 200023_s_at | 0.99998 |
| Eif3h | 230570_at | 0.999979 |
| Epcam | 201839_s_at | 0.99998 |
| Fpr2 | 210773_s_at | 0.999979 |
| Gtf3a | 201338_x_at | 0.99998 |
| Hbegf | 203821_at | 0.999981 |
| Hla-Dma | 217478_s_at | 0.99998 |
| Hla-Dpb1 | 244485_at | 0.999975 |
| Hla-Dqa1 | 203290_at | 0.999981 |
| Igfbp3 | 210095_s_at | 0.999974 |
| Lamtor5 | 202300_at | 0.999976 |
| Lyve1 | 219059_s_at | 0.999981 |
| Mif | 217871_s_at | 0.999981 |
| Mpnd | 233651_s_at | 0.999981 |
| Ntsr2 | 206899_at | 0.99998 |
| Rgs12 | 209639_s_at | 0.999965 |
| Rnf139 | 209510_at | 0.999981 |
| St13 | 207040_s_at | 0.99998 |
| Susd5 | 214954_at | 0.99998 |
| Tacstd2 | 202286_s_at | 0.999981 |
| Uchl1 | 201387_s_at | 0.999979 |
| Usp14 | 201672_s_at | 0.999976 |
| Vav2 | 205537_s_at | 0.999979 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardo, S.D.; Mazzon, E.; Mangano, K.; Basile, M.S.; Cavalli, E.; Mammana, S.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Transcriptomic Analysis Reveals Involvement of the Macrophage Migration Inhibitory Factor Gene Network in Duchenne Muscular Dystrophy. Genes 2019, 10, 939. https://doi.org/10.3390/genes10110939
Lombardo SD, Mazzon E, Mangano K, Basile MS, Cavalli E, Mammana S, Fagone P, Nicoletti F, Petralia MC. Transcriptomic Analysis Reveals Involvement of the Macrophage Migration Inhibitory Factor Gene Network in Duchenne Muscular Dystrophy. Genes. 2019; 10(11):939. https://doi.org/10.3390/genes10110939
Chicago/Turabian StyleLombardo, Salvo Danilo, Emanuela Mazzon, Katia Mangano, Maria Sofia Basile, Eugenio Cavalli, Santa Mammana, Paolo Fagone, Ferdinando Nicoletti, and Maria Cristina Petralia. 2019. "Transcriptomic Analysis Reveals Involvement of the Macrophage Migration Inhibitory Factor Gene Network in Duchenne Muscular Dystrophy" Genes 10, no. 11: 939. https://doi.org/10.3390/genes10110939
APA StyleLombardo, S. D., Mazzon, E., Mangano, K., Basile, M. S., Cavalli, E., Mammana, S., Fagone, P., Nicoletti, F., & Petralia, M. C. (2019). Transcriptomic Analysis Reveals Involvement of the Macrophage Migration Inhibitory Factor Gene Network in Duchenne Muscular Dystrophy. Genes, 10(11), 939. https://doi.org/10.3390/genes10110939