Transcriptional Networks of Microglia in Alzheimer’s Disease and Insights into Pathogenesis


Abstract
1. Introduction: The Role of Microglia in Alzheimer’s Disease
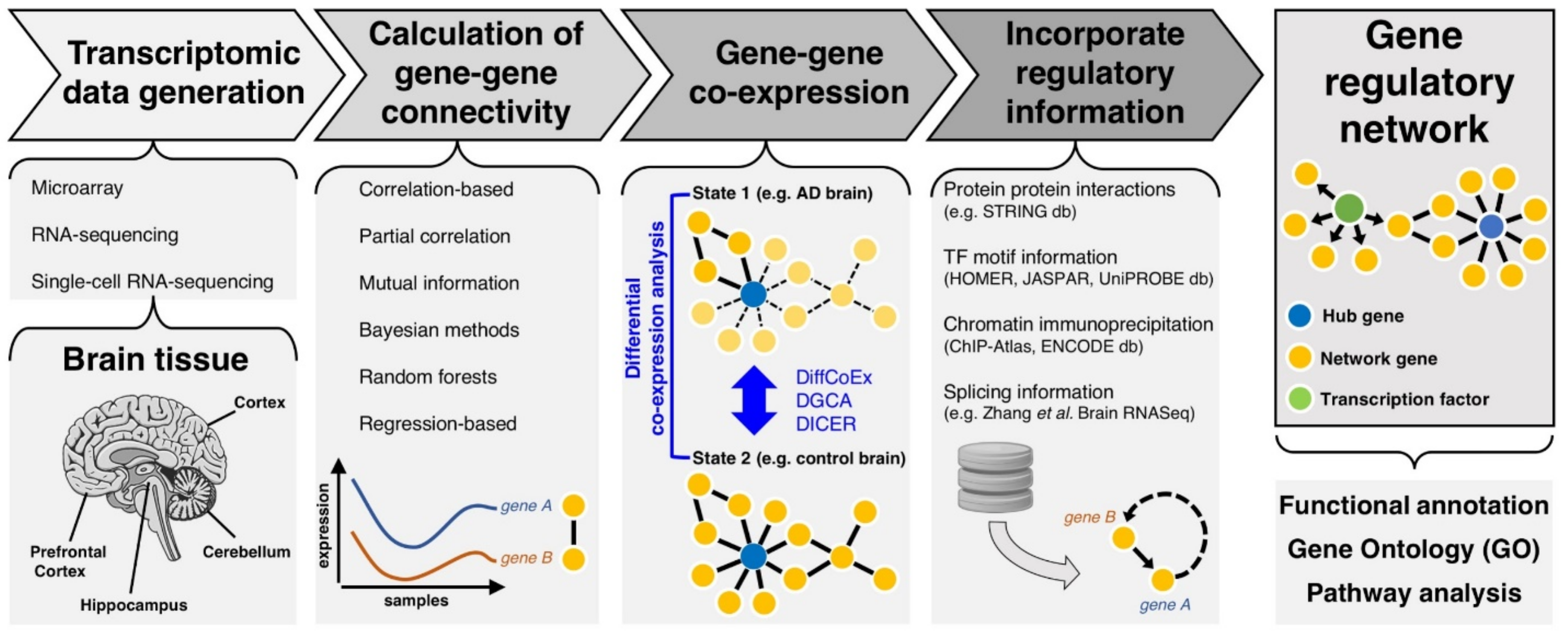
2. Transcriptomic Networks to Understand Functional Gene-Gene Interactions in AD
2.1. Primers of Transcriptional Gene Networks
2.2. From Gene Co-Expression to Regulatory Networks in Disease
3. Advancements in Transcriptomics and Gene Network Analysis
3.1. Assessing the Transcriptome
3.2. Microarray-Based Network Analyses in AD
3.3. RNA-Seq-Based Studies in AD
3.4. Single-Cell RNA-Sequencing (scRNA-sequencing) in AD
4. Further Applications of Transcriptomic Gene Networks in AD
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dementia 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar]
- Sharp, E.S.; Gatz, M. Relationship between education and dementia: An updated systematic review. Alzheimer Dis. Assoc. Disord. 2011, 25, 289–304. [Google Scholar] [CrossRef]
- Chen, N.-C.; Liang, C.-K.; Yin, C.-H.; Lin, Y.-T.; Lee, C.-C.; Chen, C.-L. Effects of Socioeconomic Status on Alzheimer Disease Mortality in Taiwan. Am. J. Geriatr. Psychiatry 2019. [Google Scholar] [CrossRef]
- Jia, R.-X.; Liang, J.-H.; Xu, Y.; Wang, Y.-Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: A meta-analysis. BMC Geriatr. 2019, 19, 181. [Google Scholar] [CrossRef]
- Larson, E.B.; Wang, L. Exercise, Aging, and Alzheimer Disease. Alzheimer Dis. Assoc. Dis. 2004, 18, 54–56. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176. [Google Scholar] [CrossRef]
- Pinheiro, L.; Faustino, C. Therapeutic Strategies Targeting Amyloid-β in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 418–452. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.G.; Kettenmann, H. Microglia in Physiology and Disease. Ann. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L. Microglia: Roles of Microglia in Chronic Neurodegenerative Diseases. In Neuroinflammation; Humana Press: Totowa, NJ, USA, 2003; pp. 3–27. [Google Scholar]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef]
- Mishra, M.K.; Wee Yong, V. Macrophages and Microglia in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Mult. Scler. Immunol. 2013. [Google Scholar] [CrossRef]
- Zhang, S. Microglial activation after ischaemic stroke. Stroke Vasc. Neurol. 2019, 4, 71–74. [Google Scholar] [CrossRef]
- Santiago, A.R.; Bernardino, L.; Agudo-Barriuso, M.; Gonçalves, J. Microglia in Health and Disease: A Double-Edged Sword. Mediators Inflamm. 2017, 2017, 7034143. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Edwards, F.A. A Unifying Hypothesis for Alzheimer’s Disease: From Plaques to Neurodegeneration. Trends Neurosci. 2019, 42, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.-C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in Alzheimer’s Disease: Risk Factors and Inflammation. Front. Neurol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017, 36, 583–603. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Esiri, M.M.; Chance, S.A. Cognitive reserve, cortical plasticity and resistance to Alzheimer’s disease. Alzheimers Res. Ther. 2012, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Reitboeck, P.; Phillips, A.; Piers, T.M.; Villegas-Llerena, C.; Butler, M.; Mallach, A.; Rodrigues, C.; Arber, C.E.; Heslegrave, A.; Zetterberg, H.; et al. Human Induced Pluripotent Stem Cell-Derived Microglia-Like Cells Harboring TREM2 Missense Mutations Show Specific Deficits in Phagocytosis. Cell Rep. 2018, 24, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176. [Google Scholar] [CrossRef] [PubMed]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Linnartz-Gerlach, B.; Bodea, L.-G.; Klaus, C.; Ginolhac, A.; Halder, R.; Sinkkonen, L.; Walter, J.; Colonna, M.; Neumann, H. TREM2 triggers microglial density and age-related neuronal loss. Glia 2019, 67, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Zhou, Y.; Ulland, T.K.; Colonna, M. TREM2-Dependent Effects on Microglia in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 202. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Tang, W.; Wang, H.; Vitek, M.P.; Bennett, E.R.; Sullivan, P.M.; Warner, D.S.; Laskowitz, D.T. APOE Genotype and an ApoE-mimetic Peptide Modify the Systemic and Central Nervous System Inflammatory Response. J. Biol. Chem. 2003, 278, 48529–48533. [Google Scholar] [CrossRef] [PubMed]
- Tansey, K.E.; Cameron, D.; Hill, M.J. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Way, J.C.; Silver, P.A. Systems engineering without an engineer: Why we need systems biology. Complexity 2007, 13, 22–29. [Google Scholar] [CrossRef]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.E.; Kontos, K.; Lafitte, F.; Bontempi, G. Information-Theoretic Inference of Large Transcriptional Regulatory Networks. EURASIP J. Bioinform. Syst. Biol. 2007, 2007, 1–9. [Google Scholar] [CrossRef]
- Altay, G.; Emmert-Streib, F. Inferring the conservative causal core of gene regulatory networks. BMC Syst. Biol. 2010, 4, 132. [Google Scholar] [CrossRef]
- Tamada, Y.; Shimamura, T.; Yamaguchi, R.; Imoto, S.; Nagasaki, M.; Miyano, S. Sign: Large-scale gene network estimation environment for high performance computing. Genome Inform. 2011, 25, 40–52. [Google Scholar]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Haury, A.-C.; Mordelet, F.; Vera-Licona, P.; Vert, J.-P. TIGRESS: Trustful Inference of Gene Regulation using Stability Selection. BMC Syst. Biol. 2012, 6, 145. [Google Scholar] [CrossRef]
- Delgado, F.M.; Gómez-Vela, F. Computational methods for Gene Regulatory Networks reconstruction and analysis: A review. Artif. Intell. Med. 2019, 95, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Regev, A.; Roy, S. Comparative analysis of gene regulatory networks: From network reconstruction to evolution. Annu. Rev. Cell Dev. Biol. 2015, 31, 399–428. [Google Scholar] [CrossRef] [PubMed]
- Tieri, P.; Farina, L.; Petti, M.; Astolfi, L.; Paci, P.; Castiglione, F. Network Inference and Reconstruction in Bioinformatics. Encycl. Bioinform. Comput. Biol. 2019, 805–813. [Google Scholar] [CrossRef]
- Schäfer, J.; Strimmer, K. An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics 2005, 21, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jhong, J.-H.; Lee, J.; Koo, J.-Y.; Lee, B.; Han, S. Node-Structured Integrative Gaussian Graphical Model Guided by Pathway Information. Comput. Math. Methods Med. 2017, 2017, 8520480. [Google Scholar] [CrossRef]
- Villa-Vialaneix, N.; Liaubet, L.; Laurent, T.; Cherel, P.; Gamot, A.; SanCristobal, M. The structure of a gene co-expression network reveals biological functions underlying eQTLs. PLoS ONE 2013, 8, e60045. [Google Scholar] [CrossRef]
- Allocco, D.J.; Kohane, I.S.; Butte, A.J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinform. 2004, 5, 18. [Google Scholar] [CrossRef]
- Josyula, N.; Andersen, M.E.; Kaminski, N.; Dere, E.; Zacharewski, T.R.; Bhattacharya, S. Gene Coregulation and Coexpression in the Aryl Hydrocarbon Receptor-mediated Transcriptional Regulatory Network in the Mouse Liver. bioRxiv 2018, 260018. [Google Scholar] [CrossRef]
- Saha, A.; Kim, Y.; Gewirtz, A.D.H.; Jo, B.; Gao, C.; McDowell, I.C. GTEx Consortium; Engelhardt, B.E.; Battle, A. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 2017, 27, 1843–1858. [Google Scholar] [CrossRef]
- Yeung, K.Y.; Medvedovic, M.; Bumgarner, R.E. From co-expression to co-regulation: How many microarray experiments do we need? Genome Biol. 2004, 5, R48. [Google Scholar] [CrossRef]
- Ding, J.; Kong, W.; Mou, X.; Wang, S. Construction of Transcriptional Regulatory Network of Alzheimer’s Disease Based on PANDA Algorithm. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.M.; De Bastiani, M.A.; Zimmer, E.R.; Klamt, F. Alzheimer’s disease master regulators analysis: Search for potential molecular targets and drug repositioning candidates. Alzheimer Res. Ther. 2018, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Chumakov, S.; Ballesteros, E.; Rodriguez Sanchez, J.E.; Chavez, A.; Zhang, M.; Pettit, B.M.; Fofanov, Y. Using Mutual Information to Discover Temporal Patterns in Gene Expression Data. Med. Phys. Mex. Symp. Med. Phys. 2006, 854, 25–30. [Google Scholar] [PubMed]
- Liu, B.-H. Differential Coexpression Network Analysis for Gene Expression Data. Methods Mol. Biol. 2018, 1754, 155–165. [Google Scholar]
- Tesson, B.M.; Breitling, R.; Jansen, R.C. DiffCoEx: A simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinform. 2010, 11, 497. [Google Scholar] [CrossRef]
- Amar, D.; Safer, H.; Shamir, R. Dissection of regulatory networks that are altered in disease via differential co-expression. PLoS Comput. Biol. 2013, 9, e1002955. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Z.-P. Detecting Diagnostic Biomarkers of Alzheimer’s Disease by Integrating Gene Expression Data in Six Brain Regions. Front. Genet. 2019, 10, 157. [Google Scholar] [CrossRef]
- Yu, D.; Lim, J.; Wang, X.; Liang, F.; Xiao, G. Enhanced construction of gene regulatory networks using hub gene information. BMC Bioinform. 2017, 18, 186. [Google Scholar] [CrossRef]
- Meng, G.; Mei, H. Transcriptional Dysregulation Study Reveals a Core Network Involving the Progression of Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 101. [Google Scholar] [CrossRef]
- Wehrspaun, C.C.; Haerty, W.; Ponting, C.P. Microglia recapitulate a hematopoietic master regulator network in the aging human frontal cortex. Neurobiol. Aging 2015, 36, 2443-e9. [Google Scholar] [CrossRef]
- Yao, L.; Kan, E.M.; Kaur, C.; Dheen, S.T.; Hao, A.; Lu, J.; Ling, E.-A. Notch-1 signaling regulates microglia activation via NF-κB pathway after hypoxic exposure in vivo and in vitro. PLoS ONE 2013, 8, e78439. [Google Scholar] [CrossRef]
- Yang, X.; He, G.; Hao, Y.; Chen, C.; Li, M.; Wang, Y.; Zhang, G.; Yu, Z. The role of the JAK2-STAT3 pathway in pro-inflammatory responses of EMF-stimulated N9 microglial cells. J. Neuroinflamm. 2010, 7, 54. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef]
- Blalock, E.M.; Buechel, H.M.; Popovic, J.; Geddes, J.W.; Landfield, P.W. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J. Chem. Neuroanat. 2011, 42, 118–126. [Google Scholar] [CrossRef]
- Colangelo, V.; Schurr, J.; Ball, M.J.; Pelaez, R.P.; Bazan, N.G.; Lukiw, W.J. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: Transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 2002, 70, 462–473. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Hemby, S.E.; Lee, V.M.; Eberwine, J.H.; Trojanowski, J.Q. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann. Neurol. 2000, 48, 77–87. [Google Scholar] [CrossRef]
- Liang, W.S.; Dunckley, T.; Beach, T.G.; Grover, A.; Mastroeni, D.; Ramsey, K.; Caselli, R.J.; Kukull, W.A.; McKeel, D.; Morris, J.C.; et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: A reference data set. Physiol. Genom. 2008, 33, 240–256. [Google Scholar] [CrossRef]
- Yao, P.J.; Zhu, M.; Pyun, E.I.; Brooks, A.I.; Therianos, S.; Meyers, V.E.; Coleman, P.D. Defects in expression of genes related to synaptic vesicle traffickingin frontal cortex of Alzheimer’s disease. Neurobiol. Dis. 2003, 12, 97–109. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Bonchev, D. Network Topology Analysis of Post-Mortem Brain Microarrays Identifies More Alzheimer’s Related Genes and MicroRNAs and Points to Novel Routes for Fighting with the Disease. PLoS ONE 2016, 11, e0144052. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.K.; Singh, S.M. Hippocampal gene expression meta-analysis identifies aging and age-associated spatial learning impairment (ASLI) genes and pathways. PLoS ONE 2013, 8, e69768. [Google Scholar] [CrossRef]
- Allison, S.L.; Fagan, A.M.; Morris, J.C.; Head, D. Spatial Navigation in Preclinical Alzheimer’s Disease. J. Alzheimer Dis. 2016, 52, 77–90. [Google Scholar] [CrossRef]
- Yin, J.-X.; Turner, G.H.; Lin, H.-J.; Coons, S.W.; Shi, J. Deficits in spatial learning and memory is associated with hippocampal volume loss in aged apolipoprotein E4 mice. J. Alzheimers Dis. 2011, 27, 89–98. [Google Scholar] [CrossRef]
- Uddin, R.; Singh, S.M. Gene Network Construction from Microarray Data Identifies a Key Network Module and Several Candidate Hub Genes in Age-Associated Spatial Learning Impairment. Front. Syst. Neurosci. 2017, 11, 75. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Miller, J.A.; Oldham, M.C.; Geschwind, D.H. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J. Neurosci. 2008, 28, 1410–1420. [Google Scholar] [CrossRef]
- Miller, J.A.; Horvath, S.; Geschwind, D.H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 12698–12703. [Google Scholar] [CrossRef]
- Castillo, E.; Leon, J.; Mazzei, G.; Abolhassani, N.; Haruyama, N.; Saito, T.; Saido, T.; Hokama, M.; Iwaki, T.; Ohara, T.; et al. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 2017, 7, 17762. [Google Scholar] [CrossRef]
- Mukherjee, S.; Klaus, C.; Pricop-Jeckstadt, M.; Miller, J.A.; Struebing, F.L. A Microglial Signature Directing Human Aging and Neurodegeneration-Related Gene Networks. Front. Neurosci. 2019, 13, 2. [Google Scholar] [CrossRef]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef]
- Haure-Mirande, J.-V.; Audrain, M.; Fanutza, T.; Kim, S.H.; Klein, W.L.; Glabe, C.; Readhead, B.; Dudley, J.T.; Blitzer, R.D.; Wang, M.; et al. Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer’s pathology. Acta Neuropathol. 2017, 134, 769–788. [Google Scholar] [CrossRef]
- Datta, M.; Staszewski, O.; Raschi, E.; Frosch, M.; Hagemeyer, N.; Tay, T.L.; Blank, T.; Kreutzfeldt, M.; Merkler, D.; Ziegler-Waldkirch, S.; et al. Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity 2018, 48, 514–529. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef]
- Freilich, R.W.; Woodbury, M.E.; Ikezu, T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS ONE 2013, 8, e79416. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- Iancu, O.D.; Colville, A.; Oberbeck, D.; Darakjian, P.; McWeeney, S.K.; Hitzemann, R. Cosplicing network analysis of mammalian brain RNA-Seq data utilizing WGCNA and Mantel correlations. Front. Genet. 2015, 6, 174. [Google Scholar] [CrossRef]
- Dai, C.; Li, W.; Liu, J.; Zhou, X.J. Integrating many co-splicing networks to reconstruct splicing regulatory modules. BMC Syst. Biol. 2012, 6, S17. [Google Scholar] [CrossRef]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.-C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Song, J. The Role of Long Noncoding RNAs in Diabetic Alzheimer’s Disease. J. Clin. Med. Res. 2018, 7, 461. [Google Scholar] [CrossRef]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.-U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Vagnoni, A.; Perkinton, M.S.; Gray, E.H.; Francis, P.T.; Noble, W.; Miller, C.C.J. Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Aβ production. Hum. Mol. Genet. 2012, 21, 2845–2854. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Yin, L.; Thambisetty, M.; Troncoso, J.C.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 2018, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef]
- Wes, P.D.; Holtman, I.R.; Boddeke, E.W.G.M.; Möller, T.; Eggen, B.J.L. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 2016, 64, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Magistri, M.; Velmeshev, D.; Makhmutova, M.; Faghihi, M.A. Transcriptomics Profiling of Alzheimer’s Disease Reveal Neurovascular Defects, Altered Amyloid-β Homeostasis, and Deregulated Expression of Long Noncoding RNAs. J. Alzheimer Dis. 2015, 48, 647–665. [Google Scholar] [CrossRef]
- Hu, G.; Liao, K.; Niu, F.; Yang, L.; Dallon, B.W.; Callen, S.; Tian, C.; Shu, J.; Cui, J.; Sun, Z.; et al. Astrocyte EV-Induced lincRNA-Cox2 Regulates Microglial Phagocytosis: Implications for Morphine-Mediated Neurodegeneration. Mol. Ther. Nucl. Acids 2018, 13, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Litvinchuk, A.; Wan, Y.-W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.-Y.; Lee, V.M. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.-A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Liu, J.; Lin, D.; Zhang, J.; Terrando, N.; Wu, A. Complement activation contributes to perioperative neurocognitive disorders in mice. J. Neuroinflamm. 2018, 15, 254. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, X.; Chen, C.; Yuan, W.; Wang, X.; Li, M.; Yu, Z. STAT3 signalling pathway is involved in the activation of microglia induced by 2.45 GHz electromagnetic fields. Int. J. Radiat. Biol. 2010, 86, 27–36. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Ryder, J.W.; Hole, J.T.; Ebert, P.J.; Airey, D.C.; Qian, H.-R.; Logsdon, B.; Fisher, A.; Ahmed, Z.; et al. Genome-wide RNAseq study of the molecular mechanisms underlying microglia activation in response to pathological tau perturbation in the rTg4510 tau transgenic animal model. Mol. Neurodegener. 2018, 13, 65. [Google Scholar] [CrossRef]
- Wes, P.D.; Easton, A.; Corradi, J.; Barten, D.M.; Devidze, N.; DeCarr, L.B.; Truong, A.; He, A.; Barrezueta, N.X.; Polson, C.; et al. Tau Overexpression Impacts a Neuroinflammation Gene Expression Network Perturbed in Alzheimer’s Disease. PLoS ONE 2014, 9, e106050. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.-V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Birnbaum, K.D. Power in Numbers: Single-Cell RNA-Seq Strategies to Dissect Complex Tissues. Annu. Rev. Genet. 2018, 52, 203–221. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef]
- Todorov, H.; Cannoodt, R.; Saelens, W.; Saeys, Y. Network Inference from Single-Cell Transcriptomic Data. Methods Mol. Biol. 2019, 1883, 235–249. [Google Scholar]
- Xu, R.; Nettleton, D.; Nordman, D.J. Case-Specific Random Forests. J. Comput. Graph. Stat. 2016, 25, 49–65. [Google Scholar] [CrossRef]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kiryu, H.; Furusawa, C.; Ko, M.S.H.; Ko, S.B.H.; Gouda, N.; Hayashi, T.; Nikaido, I. SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017, 33, 2314–2321. [Google Scholar] [CrossRef]
- Lummertz da Rocha, E.; Rowe, R.G.; Lundin, V.; Malleshaiah, M.; Jha, D.K.; Rambo, C.R.; Li, H.; North, T.E.; Collins, J.J.; Daley, G.Q. Reconstruction of complex single-cell trajectories using CellRouter. Nat. Commun. 2018, 9, 892. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Liu, W.; Taso, O.; Wang, R.; Garcia-Reitboeck, P.; Andrews, W.D.; Piers, T.M.; Pocock, J.M.; Cummings, D.M.; Hardy, J.; Edwards, F.A.; et al. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. bioRxiv 2018, 449884. [Google Scholar]
- Iacono, G.; Massoni-Badosa, R.; Heyn, H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019, 20, 110. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada Gonzalez, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Zeisel, A.; Munoz-Manchado, A.B.; Codeluppi, S.; Lonnerberg, P.; La Manno, G.; Jureus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.-H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015, 518, 365–369. [Google Scholar] [CrossRef]
- Suo, S.; Zhu, Q.; Saadatpour, A.; Fei, L.; Guo, G.; Yuan, G.-C. Revealing the Critical Regulators of Cell Identity in the Mouse Cell Atlas. Cell Rep. 2018, 25, 1436–1445. [Google Scholar] [CrossRef]
- Tay, T.L.; Dautzenberg, J.; Grün, D.; Prinz, M. Unique microglia recovery population revealed by single-cell RNAseq following neurodegeneration. Acta Neuropathol. Commun. 2018, 6, 87. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Chavez-Alvarez, R.; Chavoya, A.; Mendez-Vazquez, A. Discovery of possible gene relationships through the application of self-organizing maps to DNA microarray databases. PLoS ONE 2014, 9, e93233. [Google Scholar] [CrossRef]
- Patir, A.; Shih, B.; McColl, B.W.; Freeman, T.C. A core transcriptional signature of human microglia: Derivation and utility in describing region-dependent alterations associated with Alzheimer’s disease. Glia 2019, 67, 1240–1253. [Google Scholar] [CrossRef]
- Freeman, T.C.; Goldovsky, L.; Brosch, M.; van Dongen, S.; Mazière, P.; Grocock, R.J.; Freilich, S.; Thornton, J.; Enright, A.J. Construction, visualisation, and clustering of transcription networks from microarray expression data. PLoS Comput. Biol. 2007, 3, 2032–2042. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507. [Google Scholar] [CrossRef]
- Li, T.; Kim, A.; Rosenbluh, J.; Horn, H.; Greenfeld, L.; An, D.; Zimmer, A.; Liberzon, A.; Bistline, J.; Natoli, T.; et al. GeNets: A unified web platform for network-based genomic analyses. Nat. Methods 2018, 15, 543–546. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; David Sweatt, J.; Rumbaugh, G. Inhibitors of Class 1 Histone Deacetylases Reverse Contextual Memory Deficits in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef]
- Cheray, M.; Joseph, B. Epigenetics Control Microglia Plasticity. Front. Cell. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Smith, A.M.; Smyth, L.C.; Jansson, D.; Scotter, E.L.; Swanson, M.E.V.; Aalderink, M.; Coppieters, N.; Narayan, P.; Handley, R.; et al. 1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol. Neurodegener. 2018, 13, 44. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.-M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.-C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Canchi, S.; Raao, B.; Masliah, D.; Rosenthal, S.B.; Sasik, R.; Fisch, K.M.; De Jager, P.L.; Bennett, D.A.; Rissman, R.A. Integrating Gene and Protein Expression Reveals Perturbed Functional Networks in Alzheimer’s Disease. Cell Rep. 2019, 28, 1103–1116. [Google Scholar] [CrossRef]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Duclot, F.; Kabbaj, M. The Role of Early Growth Response 1 (EGR1) in Brain Plasticity and Neuropsychiatric Disorders. Front. Behav. Neurosci. 2017, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Marttinen, M.; Paananen, J.; Neme, A.; Mitra, V.; Takalo, M.; Natunen, T.; Paldanius, K.M.A.; Mäkinen, P.; Bremang, M.; Kurki, M.I.; et al. A multiomic approach to characterize the temporal sequence in Alzheimer’s disease-related pathology. Neurobiol. Dis. 2019, 124, 454–468. [Google Scholar] [CrossRef]
- Roussarie, J.-P.; Yao, V.; Plautz, Z.; Kasturia, S.; Albornoz, C.; Schmidt, E.F.; Brichta, L.; Barnea-Cramer, A.; Heintz, N.; Hof, P.; et al. Selective neuronal vulnerability in Alzheimer’s disease: A network-based analysis. bioRxiv 2018, 499897. [Google Scholar]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Floden, A.M.; Li, S.; Combs, C.K. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J. Neurosci. 2005, 25, 2566–2575. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.-C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daniel Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef]
- Bernhardi, R.V.O.N.; Von Bernhardi, R.; Ramirez, G. Microglia–Astrocyte interaction in Alzheimer’s disease: Friends or foes for the nervous system? Biol. Res. 2001, 34, 123–128. [Google Scholar] [CrossRef]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia–Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Insights into AD Pathology | Technology | Species - Tissue type | Network Inference Method | References |
|---|---|---|---|---|
| -Upregulation of neural signaling elements and pro-inflammatory elements | Microarray | Human - Hippocampal CA1 | Cluster analysis | [78] |
| -CD4, DCN, and IL8 extracellular ligands linked to disease initiation -Implication of miRNA-networks in AD pathogenesis | Microarray | Human - Hippocampal CA1 - Entorhinal Cortex | Network Topology Analysis | [82] |
| -PSEN1 is strongly associated with myelin proteins -Conservation of modules for metabolism and synaptic plasticity conserved between AD and aging | Microarray | Human - Hippocampal CA1 | WGCNA | [88] |
| -Apoe implicated as a general aging gene and associated with syndromic learning impairment | Microarray | Rat - Hippocampus | Ingenuity Pathway Analysis (IPA) | [83] |
| -Cdk5r1, Dlg3, Kcnab2, and Mapk1, and Camk1g identified as hub network genes and associated with ion signaling and learning | Microarray | Rat - Hippocampus | WGCNA | [86] |
| -TYROBP, PTPRC, ITGB2, and Trem2 identified as “hub” genes in AD gene networks in humans and mice, respectively -Conservation of genes across humans and mice AD | Microarray RNA-Seq | Human - Prefrontal Cortex - Substantia Nigra Mouse - Hippocampus | WGCNA | [91,92] |
| -Role of splicing quantitative trait loci and co-splicing gene networks in AD -CLU, PICALM, and PTK2B show unique splicing mechanisms in AD | RNA-Seq | Human - Dorsolateral Prefrontal Cortex | WGCNA GeNets | [101,148] |
| -PARK2 associated with NLRP3 inflammasome in microglia activation - 6 FDA-approved drugs (Cefuroxime, Cyproterone, Dydrogesterone, Metrizamide, Trimethadione, and Vorinostat) predicted to modulate microglia master regulators | Microarray | Human - Hippocampus | ARACNE (Algorithm for the Reconstruction of Accurate Cellular Networks) | [63,64] |
| -Role of splicing gene networks of microglia in AD, and identification of App and Clstn1 as differentially spliced -Splicing occurs differentially across different cell types in AD brain | RNA-Seq | Mouse - Cortex | WGCNA | [105] |
| -Alternative exon-exon junction splicing in AD brain | RNA-Seq | Human - Dorsolateral Prefrontal Cortex | WGCNA | [107] |
| -C3-C3ar signaling associated with viral-synapse loss and reactive astrocytes, and its downregulation reduces tau pathology -Spi1, Trem2, and Ms4a6a part of C3ar gene network | RNA-Seq | Human - Parahippocampal Gyrus Mice - Hippocampus | Correlation-based | [112] |
| -TYROBP identified as a key regulator in a microglia module controlling phagocytosis -Different brain regions have different regulators -Functional gene networks identified for prefrontal cortex including complements and cytokine networks | RNA-Seq | Human - Dorsolateral Prefrontal Cortex - Visual Cortex - Cerebellum | Module Differential Connectivity (MDC) Causal probabilistic Bayesian | [120] |
| -Dissection of Damage-Associated Microglia(DAM) into pro-inflammatory and anti-inflammatory modules | Microarray Nanostring | Mouse - Whole brain | WGCNA | [62] |
| -Trem2 network becomes sparser with AD pathogenesis; specifically, genes that lost connectivity overlap with Trem1-dependent genes in monocytes | scRNA-seq | Mouse - Cortex - Hippocampus | Correlation-based | [135] |
| -Identification of master microglia gene regulators including PU.1/Ets family of TFs, Nfkb, Irf, and AP-1/Maf -Microglia gene network upregulation in AD | scRNA-seq | Mouse - Cortex - Hippocampus | SCENIC (Single Cell Regulatory Network Inference and Clustering) | [132] |
| -Identification of microglia immune network containing Mafb, Irf2, and Nfkb1 | scRNA-seq | Mouse - Brain | SCENIC | [139] |
| -APOE, TREM2, MEF2C, and PICALM implicated in microglia gene-trait correlation modules | scRNA-seq | Human - Prefrontal Cortex | SOM (Self Organizing Maps) | [142] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chew, G.; Petretto, E. Transcriptional Networks of Microglia in Alzheimer’s Disease and Insights into Pathogenesis. Genes 2019, 10, 798. https://doi.org/10.3390/genes10100798
Chew G, Petretto E. Transcriptional Networks of Microglia in Alzheimer’s Disease and Insights into Pathogenesis. Genes. 2019; 10(10):798. https://doi.org/10.3390/genes10100798
Chicago/Turabian StyleChew, Gabriel, and Enrico Petretto. 2019. "Transcriptional Networks of Microglia in Alzheimer’s Disease and Insights into Pathogenesis" Genes 10, no. 10: 798. https://doi.org/10.3390/genes10100798
APA StyleChew, G., & Petretto, E. (2019). Transcriptional Networks of Microglia in Alzheimer’s Disease and Insights into Pathogenesis. Genes, 10(10), 798. https://doi.org/10.3390/genes10100798