Nonclonal Chromosome Aberrations and Genome Chaos in Somatic and Germ Cells from Patients and Survivors of Hodgkin Lymphoma

 ,
, 
Abstract
1. Introduction
2. Hodgkin Lymphoma Outline
3. Genotoxicity of the Anticancer Treatment in Hodgkin Lymphoma
4. Risk for a New Cancer in Hodgkin Lymphoma Patients
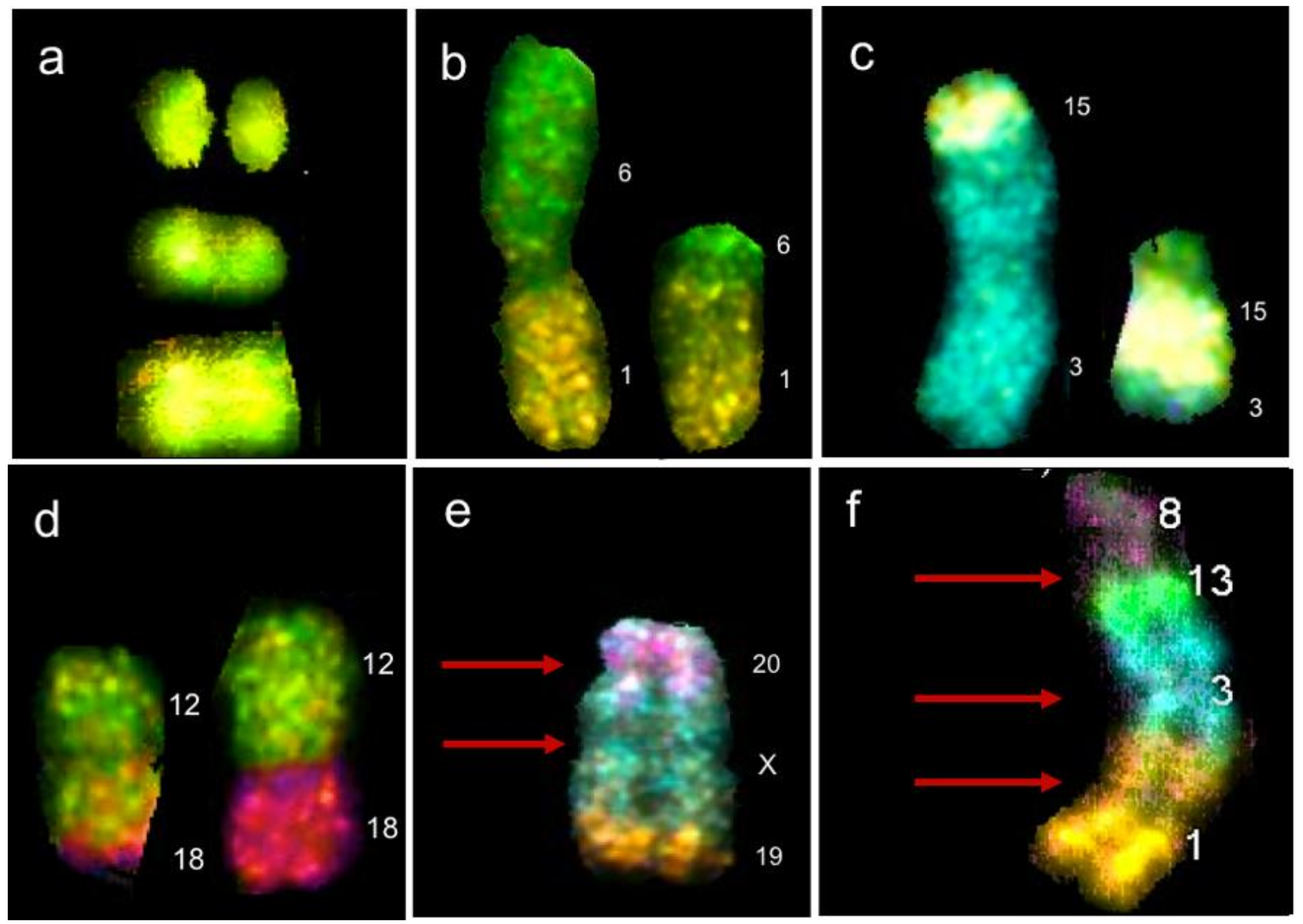
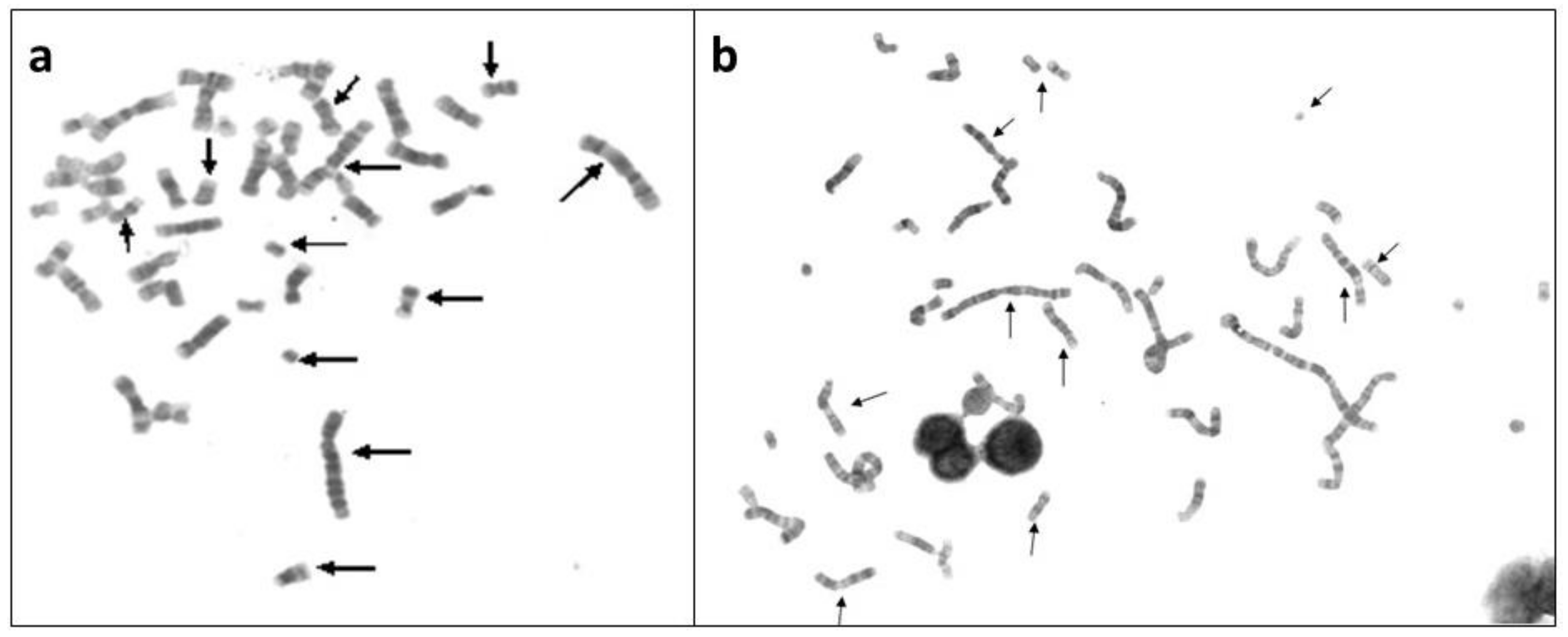
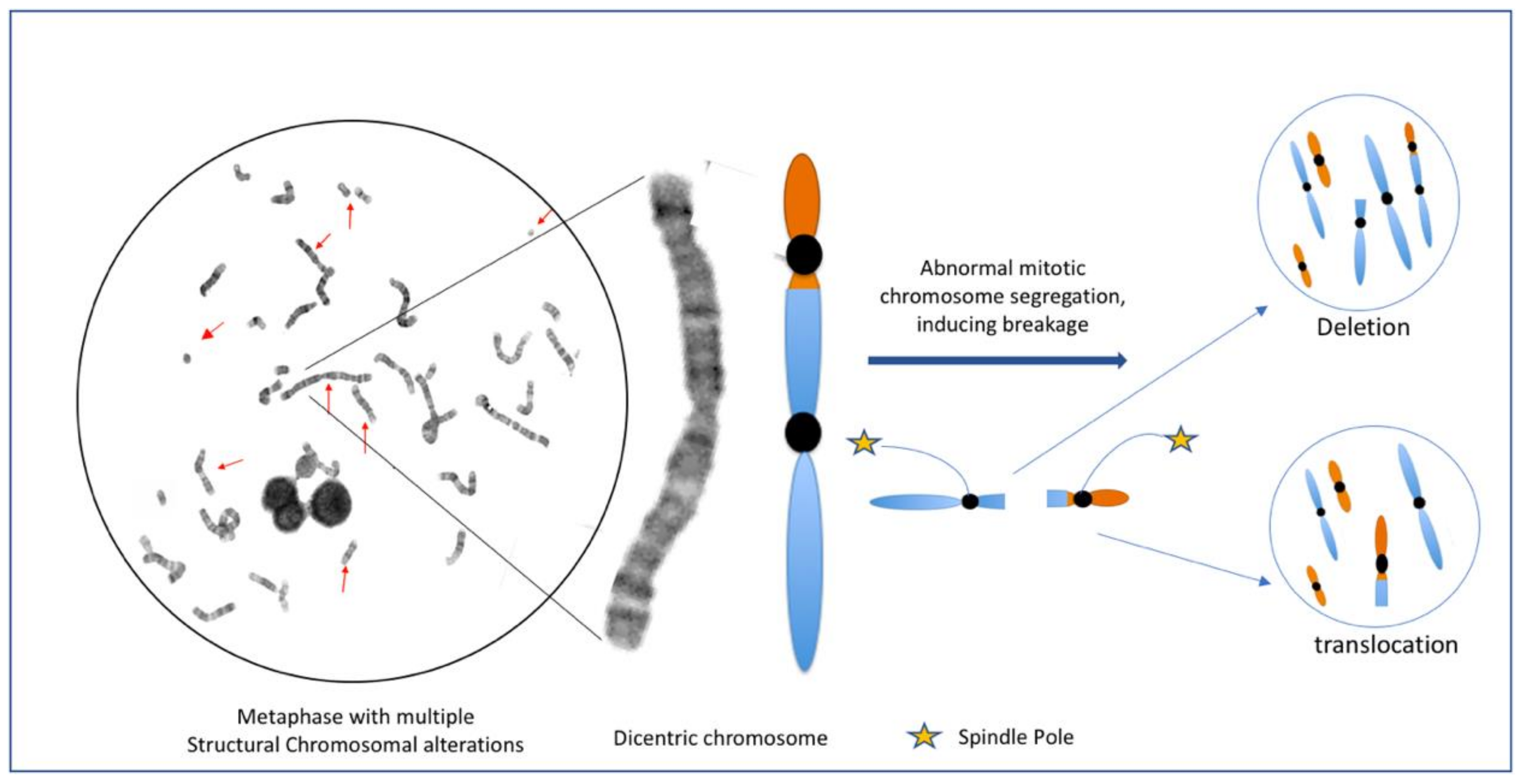
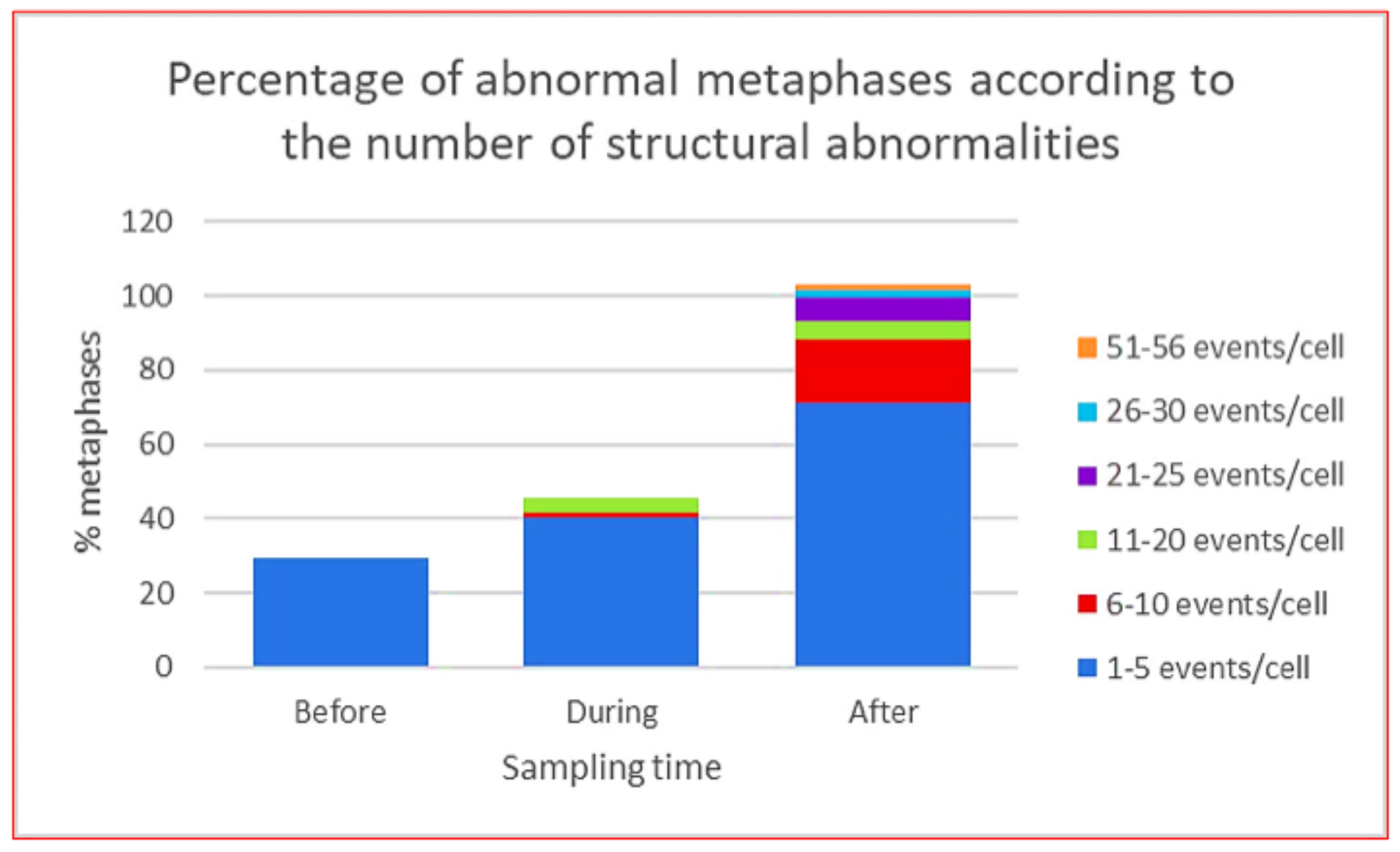
5. Genomic Instability, Chromosome Instability and Genomic Chaos
6. Impact of Anticancer Therapy in Noncancerous Somatic Cells from Patients with Hodgkin Lymphoma
7. Role of Virus Activation in the Generation of Chromosomal Instability in Somatic Noncancerous Cells
8. Impact of Anticancer Therapy on Fertility and Germ Cell Genotoxicity in Patients with Hodgkin Lymphoma
9. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Engert, A.; Horning, S.J. Hodgkin Lymphoma: A Comprehensive Update on Diagnostics and Clinics, 2nd ed.; Hodgkin Lymphoma: A Comprehensive Overview; Springer: New York, NY, USA, 2015; ISBN 978-3-319-12505-3. [Google Scholar]
- Ansell, S.M. Hodgkin Lymphoma: Diagnosis and Treatment. Mayo Clin. Proc. 2015, 90, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- De Fine Licht, S.; Rugbjerg, K.; Gudmundsdottir, T.; Bonnesen, T.G.; Asdahl, P.H.; Holmqvist, A.S.; Madanat-Harjuoja, L.; Tryggvadottir, L.; Wesenberg, F.; Hasle, H.; et al. Long-term inpatient disease burden in the Adult Life after Childhood Cancer in Scandinavia (ALiCCS) study: A cohort study of 21,297 childhood cancer survivors. PLoS Med. 2017, 14, e1002296. [Google Scholar] [CrossRef] [PubMed]
- Rendón-Macías, M.E.; Valencia-Ramón, E.A.; Fajardo-Gutiérrez, A.; Castro-Ríos, A. Incidence of Childhood Hodgkin Lymphoma in Mexico by Histologic Subtypes and Socioeconomic Regions. J. Pediatr. Hematol. Oncol. 2016, 38, e97–e101. [Google Scholar] [CrossRef] [PubMed]
- Kaseb, H.; Babiker, H. Cancer, Lymphoma, Hodgkin; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Weniger, M.; Barth, T.; Möller, P. Genomic Alterations in Hodgkin’s Lymphoma. Int. J. Hematol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Cuceu, C.; Hempel, M.W.; Sabatier, L.; Bosq, J.; Carde, P.; M’kacher, R. Chromosomal Instability in Hodgkin Lymphoma: An In-Depth Review and Perspectives. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Rotunno, M.; McMaster, M.L.; Boland, J.; Bass, S.; Zhang, X.; Burdett, L.; Hicks, B.; Ravichandran, S.; Luke, B.T.; Yeager, M.; et al. Whole exome sequencing in families at high risk for Hodgkin lymphoma: Identification of a predisposing mutation in the KDR gene. Haematologica 2016, 101, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Mai, S. LMP1 and Dynamic Progressive Telomere Dysfunction: A Major Culprit in EBV-Associated Hodgkin’s Lymphoma. Viruses 2017, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Bennaceur-Griscelli, A.; Girinsky, T.; Koscielny, S.; Delhommeau, F.; Dossou, J.; Violot, D.; Leclercq, E.; Courtier, M.H.; Beron-Gaillard, N.; et al. Telomere shortening and associated chromosomal instability in peripheral blood lymphocytes of patients with Hodgkin’s lymphoma prior to any treatment are predictive of second cancers. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.; Scott, D. The genetic toxicology of nitrogen and sulphur mustard. Mutat. Res. Genet. Toxicol. 1980, 75, 131–168. [Google Scholar] [CrossRef]
- Povirk, L.F.; Shuker, D.E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. Genet. Toxicol. 1994, 318, 205–226. [Google Scholar] [CrossRef]
- Hagemeister, F.; Purugganan, R.; Fuller, L.; McLaughlin, P.; Swan, F.J.; Romaguera, J.; Rodriguez, M.; Cabanillas, F. Treatment of early stages of Hodgkin’s disease with novantrone, vincristine, vinblastine, prednisone, and radiotherapy. Semin Hematol. 1995, 31, 36–43. [Google Scholar]
- Viviani, S.; Santoro, A.; Ragni, G.; Bonfante, V.; Bestetti, O.; Bonadonna, G. Gonadal toxicity after combination chemotherapy for hodgkins disease comparative results of MOPP vs ABVD. Eur. J. Cancer Clin. Oncol. 1985, 21, 601–605. [Google Scholar] [CrossRef]
- Jain, S.; Kapoor, G.; Bajpai, R. ABVD-Based Therapy for Hodgkin Lymphoma in Children and Adolescents: Lessons Learnt in a Tertiary Care Oncology Center in a Developing Country. Pediatr. Blood Cancer 2016, 63, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.M. Hodgkin lymphoma in children and adolescents: Improving the therapeutic index. Blood 2015, 126, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Behringer, K.; Josting, A.; Schiller, P.; Eich, H.T.; Bredenfeld, H.; Diehl, V.; Engert, A. Solid tumors in patients treated for Hodgkin’s disease: A report from the German Hodgkin Lymphoma Study Group. Ann. Oncol. 2004, 15, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R.; Engert, A.; Hansmann, M.-L. Hodgkin lymphoma. J. Clin. Investig. 2012, 122, 3439–3447. [Google Scholar] [CrossRef] [PubMed]
- Journy, N.; Mansouri, I.; Allodji, R.S.; Demoor-Goldschmidt, C.; Ghazi, D.; Haddy, N.; Rubino, C.; Veres, C.; Zrafi, W.S.; Rivera, S.; et al. Volume effects of radiotherapy on the risk of second primary cancers: A systematic review of clinical and epidemiological studies. Radiother. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H. The radical radiotherapy of regionally localized Hodgkin’s disease. Radiology 1962, 78, 553–561. [Google Scholar] [CrossRef]
- Yeoh, K.W.; Mikhaeel, N. Role of Radiotherapy in Modern Treatment of Hodgkin’s Lymphoma. Adv. Hematol. 2010, 2011, 258797. [Google Scholar] [CrossRef][Green Version]
- Yahalom, J. Does radiotherapy still have a place in Hodgkin lymphoma? Curr. Hematol. Malig. Rep. 2009, 4, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Prosnitz, L.R. Radiation doses following intensive chemotherapy in the treatment of hodgkin’s disease. Int. J. Radiat. Oncol. 1976, 1, 803–804. [Google Scholar] [CrossRef]
- Filippi, A.R.; Franco, P.; Ciammella, P. Role of modern radiation therapy in early stage Hodgkin’s lymphoma: A young radiation oncologists’ perspective. Rep. Pract. Oncol. Radiother. 2012, 17, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Bilban-Jakopin, C.; Bilban, M. Genotoxic effects of radiotherapy and chemotherapy on circulating lymphocytes in patients with Hodgkin’s disease. Mutat. Res. Toxicol. Environ. Mutagen. 2001, 497, 81–88. [Google Scholar] [CrossRef]
- Smith, L.M.; Evans, J.W.; Mori, M.; Brown, J.M.; Phil, D. The frequency of translocations after treatment for Hodgkin’s disease. Int. J. Radiat. Oncol. Biol. Phys. 1992, 24, 737–742. [Google Scholar] [CrossRef]
- Marín, A.; Martín, M.; Liñán, O.; Alvarenga, F.; López, M.; Fernández, L.; Büchser, D.; Cerezo, L. Bystander effects and radiotherapy. Reports Pract. Oncol. Radiother. 2014, 20, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Natarajan, A.T.; Hande, M.P. Chromosomal instability—Mechanisms and consequences. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 176–184. [Google Scholar] [CrossRef]
- Marcu, L.G. Photons–Radiobiological issues related to the risk of second malignancies. Phys. Med. Eur. J. Med. Phys. 2017, 42, 213–220. [Google Scholar] [CrossRef]
- Witt, K.L.; Bishop, J.B. Mutagenicity of anticancer drugs in mammalian germ cells. Mutat. Res. Mol. Mech. Mutagen. 1996, 355, 209–234. [Google Scholar] [CrossRef]
- Lowenthal, R.M.; Eaton, K. Toxicity of chemotherapy. Hematol. Oncol. Clin. N. Am. 1996, 10, 967–990. [Google Scholar] [CrossRef]
- Salas, C.; Niembro, A.; Lozano, V.; Gallardo, E.; Molina, B.; Sanchez, S.; Ramos, S.; Carnevale, A.; Perez-Vera, P.; Rivera-Luna, R.; et al. Persistent genomic instability in peripheral blood lymphocytes from hodgkin lymphoma survivors. Environ. Mol. Mutagen. 2012, 53, 271–280. [Google Scholar] [CrossRef]
- Van Eggermond, A.M.; Schaapveld, M.; Janus, C.P.; de Boer, J.P.; Krol, A.D.; Zijlstra, J.M.; van der Maazen, R.W.; Kremer, L.C.; van Leerdam, M.E.; Louwman, M.W.; et al. Infradiaphragmatic irradiation and high procarbazine doses increase colorectal cancer risk in Hodgkin lymphoma survivors. Br. J. Cancer 2017, 117, 306–314. [Google Scholar] [CrossRef]
- Van Leeuwen, F.E.; Ng, A.K. Long-term risk of second malignancy and cardiovascular disease after Hodgkin lymphoma treatment. Hematol. Am. Soc. Hematol. Educ. Progr. 2016, 2016, 323–330. [Google Scholar] [CrossRef]
- Salas, C.; Perez-Vera, P.; Frias, S. Genetic abnormalities in leukemia secondary to treatment in patients with Hodgkin’s disease. Rev. Investig. Clin. Transl. Investig. 2011, 63, 53–63. [Google Scholar]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Ye, K.J.; Ye, C.J. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef]
- García-Castillo, H.; Vásquez-Velásquez, A.I.; Rivera, H.; Barros-Núñez, P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: Delineation of clinical subtypes. Am. J. Med. Genet. Part A 2008, 146A, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed]
- García-De Teresa, B.; Hernández-Gómez, M.; Frías, S. DNA Damage as a Driver for Growth Delay: Chromosome Instability Syndromes with Intrauterine Growth Retardation. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Rao, C.V.; Asch, A.S.; Yamada, H.Y. Emerging links among Chromosome Instability (CIN), cancer, and aging. Mol. Carcinog. 2017, 56, 791–803. [Google Scholar] [CrossRef]
- Rangel, N.; Forero-Castro, M.; Rondon-Lagos, M. New Insights in the Cytogenetic Practice: Karyotypic Chaos, Non-Clonal Chromosomal Alterations and Chromosomal Instability in Human Cancer and Therapy Response. Genes 2017, 8. [Google Scholar] [CrossRef]
- Heng, H.H.; Liu, G.; Stevens, J.B.; Abdallah, B.Y.; Horne, S.D.; Ye, K.J.; Bremer, S.W.; Chowdhury, S.K.; Ye, C.J. Karyotype Heterogeneity and Unclassified Chromosomal Abnormalities. Cytogenet. Genome Res. 2013, 139, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Bohlander, S.K.; Kakadia, P.M. Chromosomal Instability in Cancer Cells; Michael, G.B., Ried, T., Eds.; Springer International Publishing: New York, NY, USA, 2015; ISBN 978-3-319-20290-7. [Google Scholar]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.Q.; Liu, G.; Bremer, S.; Ye, K.J.; Stevens, J.; Ye, C.J. Clonal and non-clonal chromosome aberrations and genome variation and aberration. Genome 2006, 49, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Stevens, J.B.; Horne, S.D.; Abdallah, B.Y.; Ye, K.J.; Bremer, S.W.; Ye, C.J.; Chen, D.J.; Heng, H.H. Genome chaos Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [PubMed]
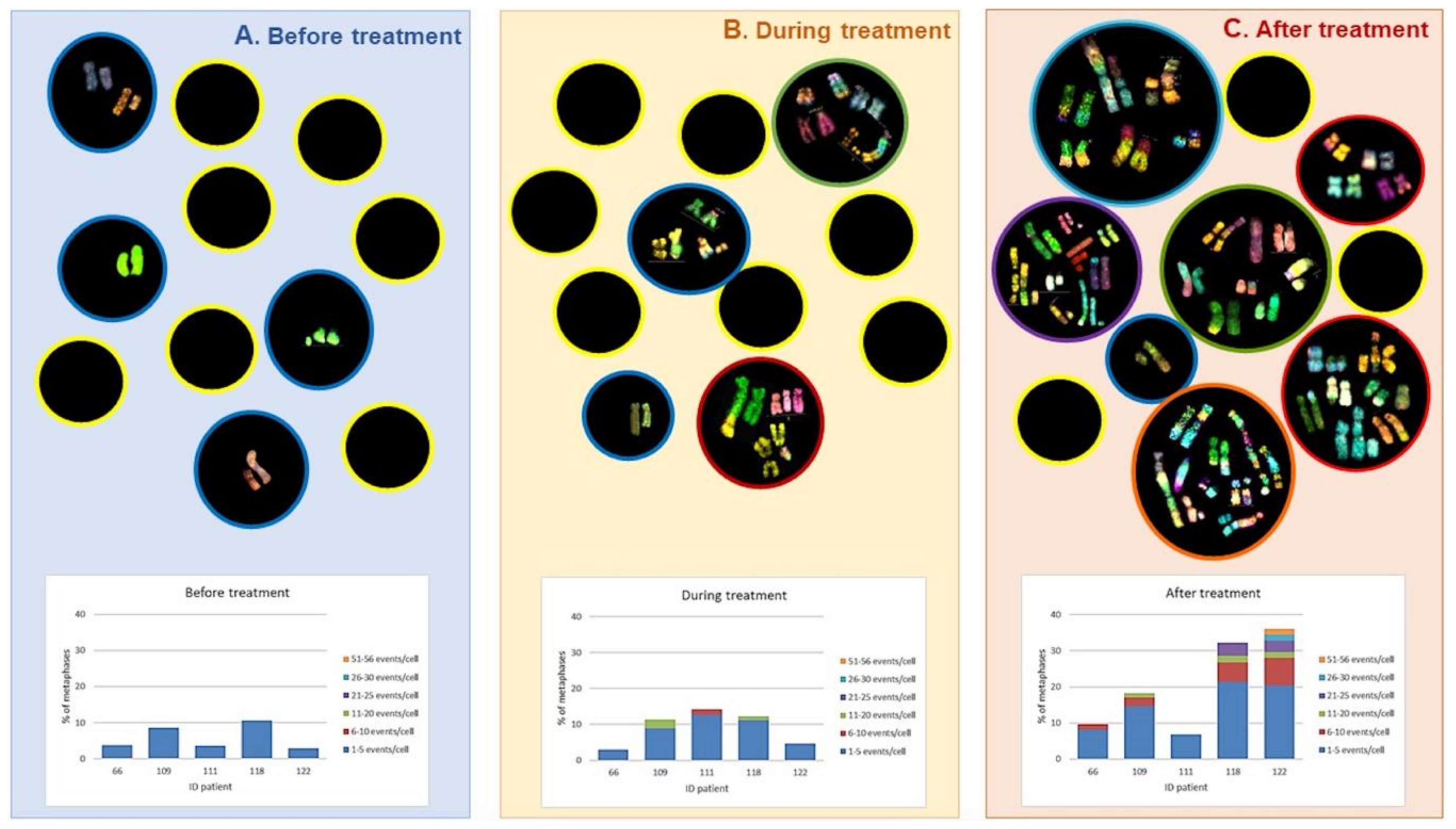
- Ramos, S.; Navarrete-Meneses, P.; Molina, B.; Cervantes-Barragán, D.E.; Lozano, V.; Gallardo, E.; Marchetti, F.; Frias, S. Genomic chaos in peripheral blood lymphocytes of Hodgkin’s lymphoma patients one year after ABVD chemotherapy/radiotherapy. Environ. Mol. Mutagen. 2018, 768. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.; Regan, S.M.; Liu, G.; Ye, C.J. Why it is crucial to analyze non clonal chromosome aberrations or NCCAs? Mol. Cytogenet. 2016, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.J.; Regan, S.; Liu, G.; Alemara, S.; Heng, H.H. Understanding aneuploidy in cancer through the lens of system inheritance, fuzzy inheritance and emergence of new genome systems. Mol. Cytogenet. 2018, 11, 31. [Google Scholar] [CrossRef]
- M’Kacher, R.; Girinsky, T.; Koscielny, S.; Dossou, J.; Violot, D.; Beron-Gaillard, N.; Ribrag, V.; Bourhis, J.; Bernheim, A.; Paramentier, C.; et al. Baseline and treatment-induced chromosomal abnormalities in peripheral blood lymphocytes of Hodgkin’s lymphoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 321–326. [Google Scholar] [CrossRef]
- De Mesa, R.L.; Sierrasesumaga, L.; Calasanz, M.J.; de Cerain, A.L.; Patino-Garcia, A. Nonclonal chromosomal aberrations induced by anti-tumoral regimens in childhood cancer: Relationship with cancer-related genes and fragile sites. Cancer Genet. Cytogenet. 2000, 121, 78–85. [Google Scholar] [CrossRef]
- Ryabchenco, N.; Nasonova, V.; Antoschina, M.; Fesenko, E.; Kondrashova, T.; Ivanova, T.; Pavlov, V.; Ryabikhina, N.; Terekhova, A. Persistence of chromosome aberrations in peripheral lymphocytes from Hodgkin’s lymphoma remission patients. Int. J. Radiat. Biol. 2003, 79, 251–257. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- M’kacher, R.; Andreoletti, L.; Flamant, S.; Milliat, F.; Girinsky, T.; Dossou, J.; Violot, D.; Assaf, E.; Clausse, B.; Koscielny, S.; et al. JC human polyomavirus is associated to chromosomal instability in peripheral blood lymphocytes of Hodgkin’s lymphoma patients and poor clinical outcome. Ann. Oncol. 2010, 21, 826–832. [Google Scholar] [CrossRef]
- Polansky, H.; Schwab, H. Latent viruses can cause disease by disrupting the competition for the limiting factor p300/CBP. Cell. Mol. Biol. Lett. 2018, 23, 56. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, A.; Uwatoko, F.; Yamamoto, M.; Tomita, K.; Majima, H.J.; Akiba, S.; Koriyama, C. Radiation-induced Epstein–Barr virus reactivation in gastric cancer cells with latent EBV infection. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Seca, H.; Brás, S.; Nascimento, M.S.J.; Vasconcelos, M.H. Treatment of Akata EBV-Positive Cells with Doxorubicin Causes More EBV Reactivation than Treatment with Etoposide. Chemotherapy 2011, 57, 195–203. [Google Scholar] [CrossRef]
- Wu, C.-C.; Liu, M.-T.; Chang, Y.-T.; Fang, C.-Y.; Chou, S.-P.; Liao, H.-W.; Kuo, K.-L.; Hsu, S.-L.; Chen, Y.-R.; Wang, P.-W.; et al. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Dutta, S.; Krajewska-Kulak, E. The Disappearing Sperms: Analysis of Reports Published Between 1980 and 2015. Am. J. Mens. Health 2017, 11, 1279–1304. [Google Scholar] [CrossRef]
- Wyrobek, A.J.; Schmid, T.E.; Marchetti, F. Relative Susceptibilities of Male Germ Cells to Genetic Defects Induced by Cancer Chemotherapies. JNCI Monogr. 2005, 2005, 31–35. [Google Scholar] [CrossRef]
- Byrne, J.; Mulvihill, J.J.; Myers, M.H.; Connelly, R.R.; Naughton, M.D.; Krauss, M.R.; Steinhorn, S.C.; Hassinger, D.D.; Austin, D.F.; Bragg, K.; et al. Effects of Treatment on Fertility in Long-Term Survivors of Childhood or Adolescent Cancer. N. Engl. J. Med. 1987, 317, 1315–1321. [Google Scholar] [CrossRef]
- Da Cunha, M.F.; Meistrich, M.L.; Fuller, L.M.; Cundiff, J.H.; Hagemeister, F.B.; Velasquez, W.S.; McLaughlin, P.; Riggs, S.A.; Cabanillas, F.F.; Salvador, P.G. Recovery of spermatogenesis after treatment for Hodgkin’s disease: Limiting dose of MOPP chemotherapy. J. Clin. Oncol. 1984, 2, 571–577. [Google Scholar] [CrossRef]
- Anselmo, A.P.; Cartoni, C.; Bellantuono, P.; Maurizi-Enrici, R.; Aboulkair, N.; Ermini, M. Risk of infertility in patients with Hodgkin’s disease treated with ABVD vs MOPP vs ABVD/MOPP. Haematologica 1990, 75, 155–158. [Google Scholar] [PubMed]
- Marmor, D.; Duyck, F. Male reproductive potential after MOPP therapy for Hodgkin’s disease: A long-term survey. Andrologia 1995, 27, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Meistrich, M.L.; Wilson, G.; Mathur, K.; Fuller, L.M.; Rodriguez, M.A.; McLaughlin, P.; Romaguera, J.E.; Cabanillas, F.F.; Ha, C.S.; Lipshultz, L.I.; et al. Rapid recovery of spermatogenesis after mitoxantrone, vincristine, vinblastine, and prednisone chemotherapy for Hodgkin’s disease. J. Clin. Oncol. 1997, 15, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Bujan, L.; Walschaerts, M.; Brugnon, F.; Daudin, M.; Berthaut, I.; Auger, J.; Saias, J.; Szerman, E.; Moinard, N.; Rives, N.; et al. Impact of lymphoma treatments on spermatogenesis and sperm deoxyribonucleic acid: A multicenter prospective study from the CECOS network. Fertil. Steril. 2014, 102, 667.e3–674.e3. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.; Walschaerts, M.; Le Mitouard, M.; Borye, R.; Thomas, C.; Auger, J.; Berthaut, I.; Brugnon, F.; Daudin, M.; Moinard, N.; et al. Impact of Hodgkin or non-Hodgkin lymphoma and their treatments on sperm aneuploidy: A prospective study by the French CECOS network. Fertil. Steril. 2017, 107, 341.e5–350.e5. [Google Scholar] [CrossRef] [PubMed]
- Canellos, G.P.; Anderson, J.R.; Propert, K.J.; Nissen, N.; Cooper, M.R.; Henderson, E.S.; Green, M.R.; Gottlieb, A.; Peterson, B.A. Chemotherapy of Advanced Hodgkin’s Disease with MOPP, ABVD, or MOPP Alternating with ABVD. N. Engl. J. Med. 1992, 327, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, S.; Molina, B.; Niembro, A.; Rivera-Luna, R.; Frias, G.; Carnevale, A.; Ulloa-Aguirre, A.; Badillo, D.; Cervantes, D.; Lozano, V.; et al. Evaluación de la función gonadal en pacientes con enfermedad de Hodgkin tratados con ABVD o MOPP. In Proceedings of the XIII Encuentro Nacional de Investigadores en Salud. Comisión Coordinadora de Institutos Nacionales de Salud y Hospitales de Alta Especialidad, Oax, México; 2008. [Google Scholar]
- Frias, S.; Van Hummelen, P.; Meistrich, M.L.; Lowe, X.R.; Hagemeister, F.B.; Shelby, M.D.; Bishop, J.B.; Wyrobek, A.J. NOVP Chemotherapy for Hodgkin’s Disease Transiently Induces Sperm Aneuploidies Associated with the Major Clinical Aneuploidy Syndromes Involving Chromosomes X, Y, 18, and 21. Cancer Res. 2003, 63, 44–51. [Google Scholar]
- Longo, D. Radiation therapy in the treatment of Hodgkin’s disease—Do you see what I see? J. Natl. Cancer Inst. 2003, 95, 928–929. [Google Scholar] [CrossRef]
- Blumenfeld, Z.; Avivi, I.; Eckman, A.; Epelbaum, R.; Rowe, J.M.; Dann, E.J. Gonadotropin-releasing hormone agonist decreases chemotherapy-induced gonadotoxicity and premature ovarian failure in young female patients with Hodgkin lymphoma. Fertil. Steril. 2008, 89, 166–173. [Google Scholar] [CrossRef]
- Behringer, K.; Thielen, I.; Mueller, H.; Goergen, H.; Eibl, A.D.; Rosenbrock, J.; Halbsguth, T.; Eichenauer, D.A.; Fuchs, M.; Reiners, K.S.; et al. Fertility and gonadal function in female survivors after treatment of early unfavorable Hodgkin lymphoma (HL) within the German Hodgkin Study Group HD14 trial. Ann. Oncol. 2012, 23, 1818–1825. [Google Scholar] [CrossRef]
- Haukvik, U.K.H.; Dieset, I.; Bjøro, T.; Holte, H.; Fosså, S.D. Treatment-related premature ovarian failure as a long-term complication after Hodgkin’s lymphoma. Ann. Oncol. 2006, 17, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Van Dorp, W.; Haupt, R.; Anderson, R.A.; Mulder, R.L.; van den Heuvel-Eibrink, M.M.; van Dulmen-den Broeder, E.; Su, H.I.; Falck Winther, J.; Hudson, M.M.; Levine, J.M.; et al. Reproductive Function and Outcomes in Female Survivors of Childhood, Adolescent, and Young Adult Cancer: A Review. J. Clin. Oncol. 2018, 36, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Brandriff, B.F.; Meistrich, M.L.; Gordon, L.A.; Carrano, A.V.; Liang, J.C. Chromosomal damage in sperm of patients surviving Hodgkin’s disease following MOPP (nitrogen mustard, vincristine, procarbazine, and prednisone) therapy with and without radiotherapy. Hum. Genet. 1994, 93, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Robbins, W.A.; Meistrich, M.L.; Moore, D.; Hagemeister, F.B.; Weier, H.-U.; Cassel, M.J.; Wilson, G.; Eskenazi, B.; Wyrobek, A.J. Chemotherapy induces transient sex chromosomal and autosomal aneuploidy in human sperm. Nat. Genet. 1997, 16, 74. [Google Scholar] [CrossRef]
- Tempest, H.G.; Ko, E.; Chan, P.; Robaire, B.; Rademaker, A.; Martin, R.H. Sperm aneuploidy frequencies analysed before and after chemotherapy in testicular cancer and Hodgkin’s lymphoma patients. Hum. Reprod. 2008, 23, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Patassini, C.; Garolla, A.; Bottacin, A.; Menegazzo, M.; Speltra, E.; Foresta, C.; Ferlin, A. Molecular Karyotyping of Human Single Sperm by Array-Comparative Genomic Hybridization. PLoS ONE 2013, 8, e60922. [Google Scholar] [CrossRef] [PubMed]
- Poot, M. Of Simple and Complex Genome Rearrangements, Chromothripsis, Chromoanasynthesis, and Chromosome Chaos. Mol. Syndromol. 2017, 8, 115–117. [Google Scholar] [CrossRef]
- Horne, S.D.; Pollick, S.A.; Heng, H.H.Q. Evolutionary mechanism unifies the hallmarks of cancer. Int. J. Cancer 2015, 136, 2012–2021. [Google Scholar] [CrossRef]
- Rigter, L.S.; Snaebjornsson, P.; Rosenberg, E.H.; Atmodimedjo, P.N.; Aleman, B.M.; ten Hoeve, J.; Geurts-Giele, W.R.; van Ravesteyn, T.W.; Hoeksel, J.; Meijer, G.A.; et al. Double somatic mutations in mismatch repair genes are frequent in colorectal cancer after Hodgkins lymphoma treatment. Gut 2018, 67, 447–455. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Drug | DNA Lesion | Altered Mechanism | Cytogenetic Alterations |
|---|---|---|---|---|
| Alkylating agents(Monofunctional and Bifunctional) | Nitrogen mustard (Mechlorethamine) Dacarbazine Procarbazine Mitomycin C Cyclophosphamide Ethyl-nitrosourea Melphalan Cisplatin Ifosphamide Clorambucil | Base damage Bulky adducts DNA intrastrand crosslinks DNA interstrand crosslink Double strand breaks | Interferes with DNA synthesis | Chromosomal deletions, insertions, inversions and translocations |
| Antibiotics | Doxorubicin (Adriamycin) Daunorubicin Epirubicin Idarubicin Bleomycin | Free radicals Crosslink DNA Single strand breaks Double strand breaks Intercalant of DNA | Blocking of DNA replication and transcription | Chromatid and chromosome-type aberrations, translocations, dicentric, acentric, and other aberrations related to damage of telomere |
| Mitosis inhibitors | Vincristine (Oncovin) Vinblastine Vinorelbine | Induce aneuploidy | Interference with tubulin polymerization and inhibits mitotic spindle | Aneuploidy and polyploidy |
| Topoisomerase II inhibitors | Daunorubicin Epirubicin Mitoxantrone (Novantrone) Camptothecin Etoposide | Single strand breaks Double strand breaks Replication lesions | Inhibition of DNA synthesis by forming a complex with Topo II and DNA | Chromosomal translocations, aneuploidy, polyploidy and endoreduplication |
| Anticancer Treatment | Pre-Treatment | Post-Treatment * | Reference | ||
|---|---|---|---|---|---|
| Normospermia (% of Patients) | Normospermia (% of Patients) | Oligospermia (% of Patients) | Azoospermia (% of Patients) | ||
| MOPP | 100 | 28 | 24 | 48 | Da Cunha et al., 1984 [63] |
| MOPP | 100 | 3 | 0 | 97 | Viviani et al., 1985 [15] |
| MOPP | 84 | 0 | 62 | 38 | Meistrich et al., 1997 [66] |
| MOPP | 100 | 20 | 35 | 45 | Sánchez et al., 2008 [70] |
| MOPP | 100 | NA | NA | 75 | Bujan et al., 2014 [67] |
| ABVD | 100 | 46 | 21 | 33 | Viviani et al., 1985 [15] |
| ABVD | 100 | 80 | 15 | 5 | Sánchez et al., 2008 [70] |
| ABVD | 100 | 100 | 0 | 0 | Bujan et al., 2014 [67] |
| NOVP | 100 | 50 | 50 | 0 | Frias et al., 2003 [71] |
| Anticancer Therapy | Chromosomal Damage | Reference (Technique) |
|---|---|---|
| Lymphocytes | ||
| MOPP/RT 6 cycles | * Chromosomal translocations, NCCA (Persistent up to 24 years) | Smith et al., 1992 [27] (FISH, painting of chromosome 4) |
| MOPP/ABV 6–9 cycles | * NCCA structural. Chromosome breaks, acentric fragments, dicentrics, and micronucleus (Persistent at six months) | Bilban-Jakopin and Bilban, 2001 [26] (Nonbanded chromosomes) |
| BEACOPP, EBVP, ABVD, MOPP/ABV RT (combination not specified) 6 cycles | * NCCA structural (Persistent up to 2 years) | M’Kacher et al., 2003 [51] (FISH, Painting of chromosomes 1, 3, and 4) |
| MOPP/RT 2–9 cycles | NCCA numerical and structural Persistent (up to 17 years) | Salas et al., 2012 [33] (G-Banding Chromosomes) |
| ABVD/RT 6–8 cycles | NCCA numerical and structural including genomic chaos (Persistent at 1 year) | Ramos et al., 2018 [48] (M-FISH) |
| Spermatozoa | ||
| MOPP/RT 2–6 cycles | * Numerical and structural NCCA (Persistent up to 20 years) | Brandriff et al.,1994 [77] (Nonbanded chromosomes) |
| CHOP/MOPP/ABV 4–7cycles | Hyperhaploidy, disomy, and diploidy (Persistent; decrease at 2 years) | Martínez et al., 2017 [68] (FISH, specific probes) |
| NOVP 3 cycles | Disomies, diploidies, and complex genotypes involving the X, Y and 8 chromosomes (Transient; decrease at 3 months) | Robbins et al., 1997 [78] (FISH, specific probes) |
| NOVP 3 cycles | Disomies, diploidies, and complex genotypes involving the X, Y and 18 and 21 chromosomes (Transient; decrease at 3 months) | Frias et al., 2003 [71] (FISH, specific probes) |
| ABVD/RT 4–8 cycles | Disomy XY, XX, Nullisomy 13 and 21 (Transient; decrease at 18 months) | Tempest et al., 2008 [79] (FISH, specific probes) |
| ABVD (number of cycles non-specified) | Disomies XY, XX, Sex chromosome nullisomy, loss and/or gain of part of chromosomes, and complex alterations | Patassini et al., 2013 [80] (microarrays aCGH) |
| ABVD/RT 4–7 cycles | Hiperhaploidy, disomy, and diploidy (Transient; decrease at 3 months) | Martínez et al., 2017 [68] (FISH, specific probes) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frias, S.; Ramos, S.; Salas, C.; Molina, B.; Sánchez, S.; Rivera-Luna, R. Nonclonal Chromosome Aberrations and Genome Chaos in Somatic and Germ Cells from Patients and Survivors of Hodgkin Lymphoma. Genes 2019, 10, 37. https://doi.org/10.3390/genes10010037
Frias S, Ramos S, Salas C, Molina B, Sánchez S, Rivera-Luna R. Nonclonal Chromosome Aberrations and Genome Chaos in Somatic and Germ Cells from Patients and Survivors of Hodgkin Lymphoma. Genes. 2019; 10(1):37. https://doi.org/10.3390/genes10010037
Chicago/Turabian StyleFrias, Sara, Sandra Ramos, Consuelo Salas, Bertha Molina, Silvia Sánchez, and Roberto Rivera-Luna. 2019. "Nonclonal Chromosome Aberrations and Genome Chaos in Somatic and Germ Cells from Patients and Survivors of Hodgkin Lymphoma" Genes 10, no. 1: 37. https://doi.org/10.3390/genes10010037
APA StyleFrias, S., Ramos, S., Salas, C., Molina, B., Sánchez, S., & Rivera-Luna, R. (2019). Nonclonal Chromosome Aberrations and Genome Chaos in Somatic and Germ Cells from Patients and Survivors of Hodgkin Lymphoma. Genes, 10(1), 37. https://doi.org/10.3390/genes10010037