RNA-Seq Analyses Identify Additivity as the Predominant Gene Expression Pattern in F1 Chicken Embryonic Brain and Liver

Abstract
1. Introduction
2. Material and Methods
2.1. Ethical Statement
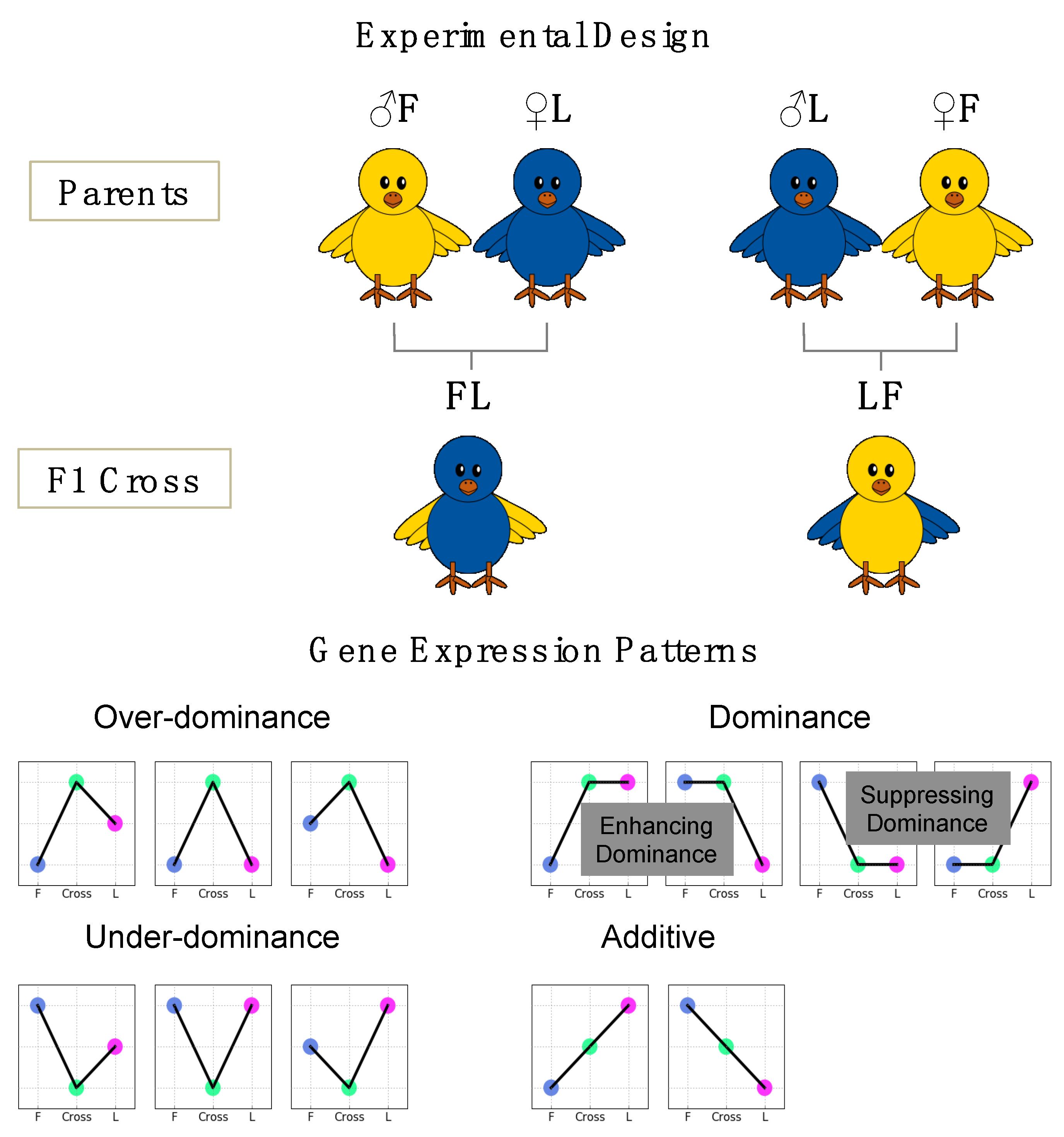
2.2. Animals
2.3. Analysis of Sequencing Data
2.4. Functional Analysis of Differentially Expressed Genes
3. Results and Discussion
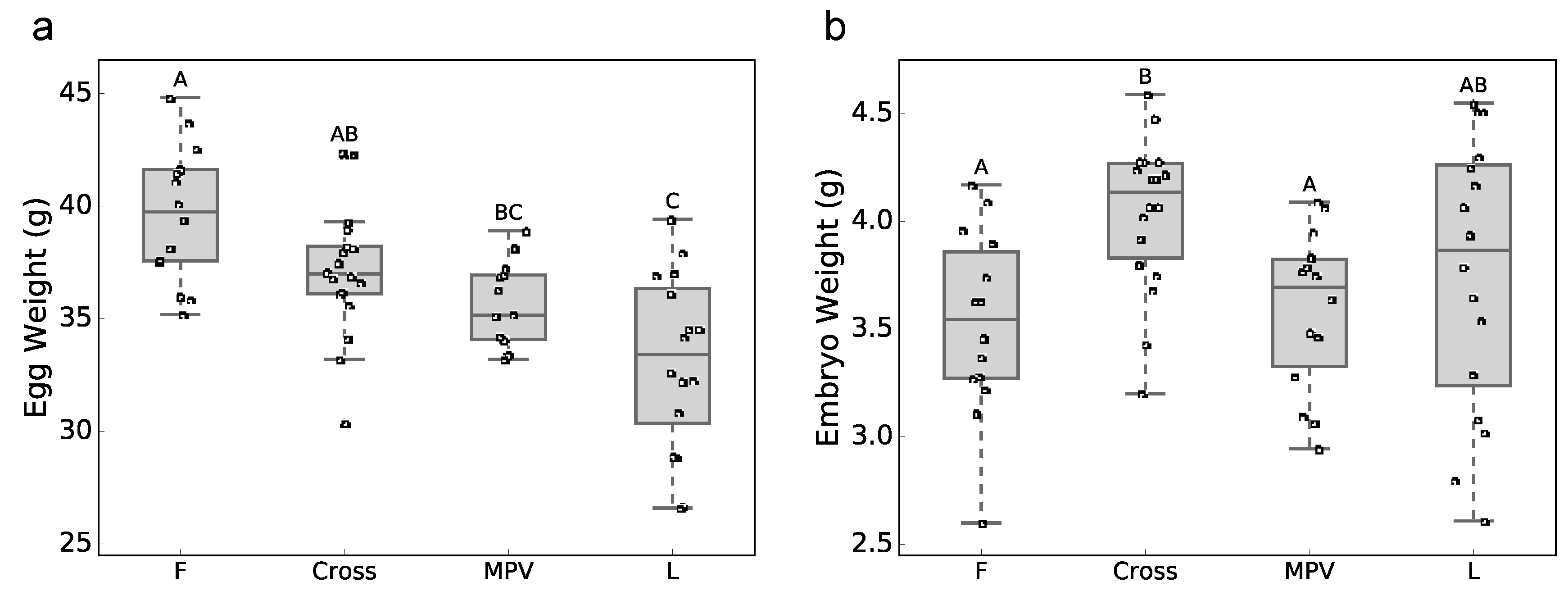
3.1. Phenotypic Data
3.2. Differentially Expressed Genes between Inbred Fayoumi and Leghorn Lines
3.3. Gene Expression of the Reciprocal Crosses
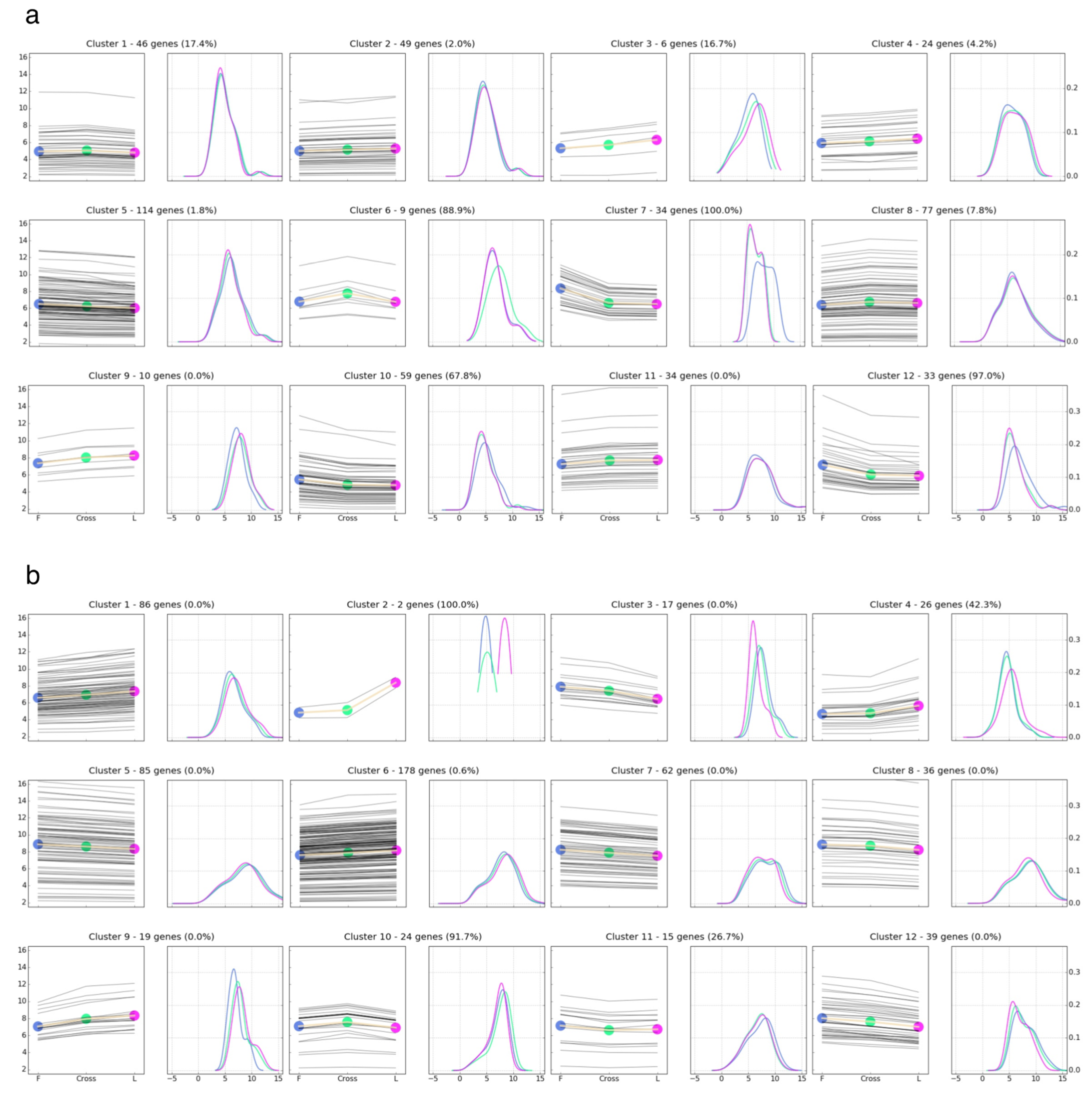
3.4. Comparison of Gene Expression Pattern across Parental Lines and F1 Crosses
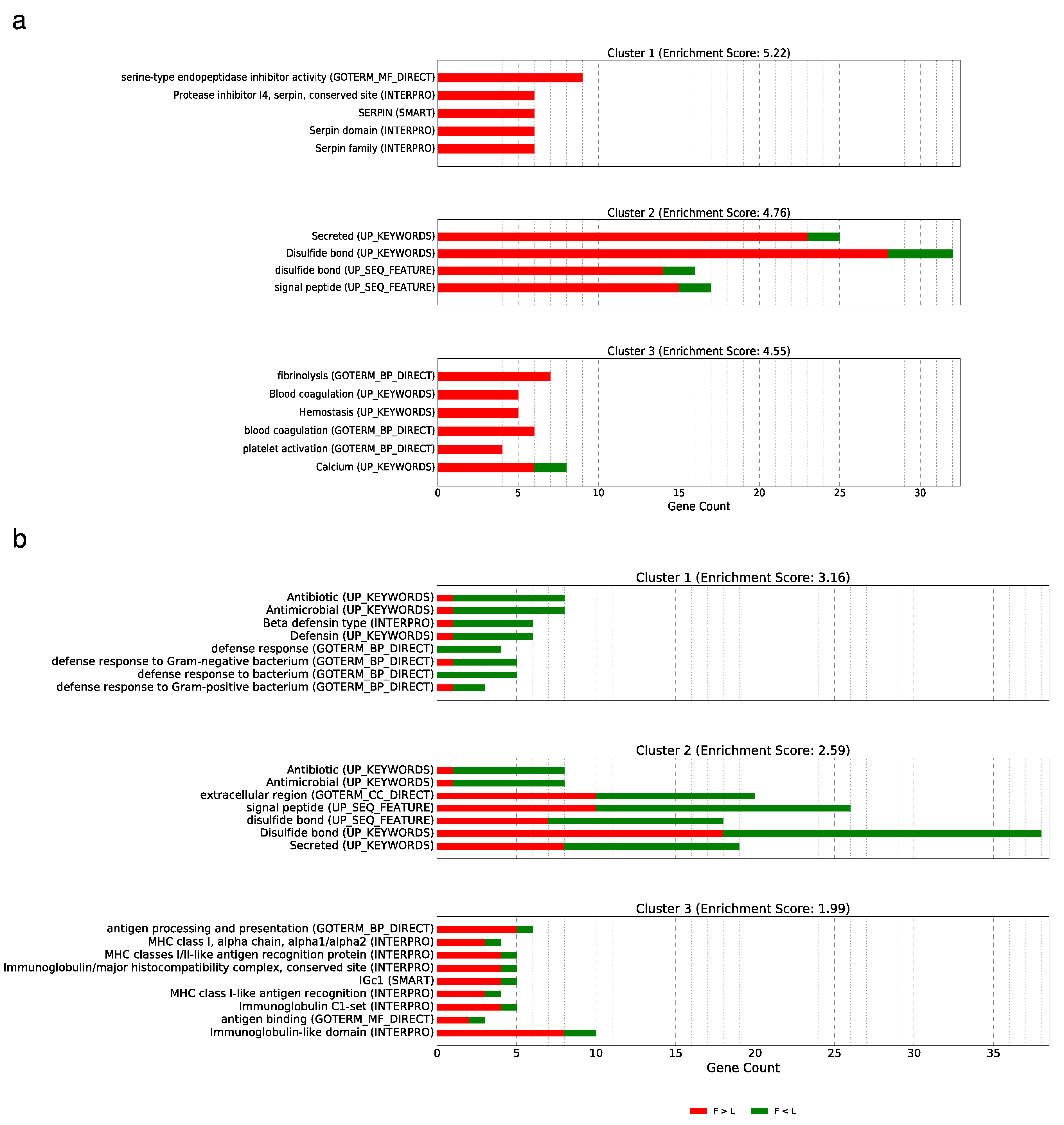
3.5. Implications and Functional Annotation of the Differentially Expressed Genes Showing a Non-Additive or Additive Expression Pattern
3.5.1. Over-Dominance
3.5.2. Dominance
3.5.3. Additive
3.6. Cis- vs. Trans-Regulation of Gene Expression
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruce, A.B. The Mendelian theory of heredity and the augmentation of vigor. Science 1910, 32, 627–628. [Google Scholar] [CrossRef]
- Jones, D.F. Dominance of linked factors as a means of accounting for heterosis. Genetics 1917, 2, 466–479. [Google Scholar] [CrossRef]
- Shull, G.H. The composition of a field of maize. J. Hered. 1908, 4, 296–301. [Google Scholar] [CrossRef]
- East, E.M. Inbreeding in corn. Con. Agric. Exp. Stn. Rep. 1908, 1907, 419–428. [Google Scholar]
- Bateson, W. Mendel’s Principles of Heredity; Cambridge University Press: Cambridge, UK, 1909. [Google Scholar]
- Fisher, R.A. XV.—The Correlation between relatives on the supposition of mendelian inheritance. Trans. R. Soc. Edinb. 1919, 52, 399–433. [Google Scholar] [CrossRef]
- Hubner, N.; Wallace, C.A.; Zimdahl, H.; Petretto, E.; Schulz, H.; Maciver, F.; Mueller, M.; Hummel, O.; Monti, J.; Zidek, V.; et al. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat. Genet. 2005, 37, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Stupar, R.M. Allelic variation and heterosis in maize: How do two halves make more than a whole? Genome Res. 2007, 17, 264–275. [Google Scholar] [CrossRef]
- Zhai, R.; Feng, Y.; Wang, H.; Zhan, X.; Shen, X.; Wu, W.; Zhang, Y.; Chen, D.; Dai, G.; Yang, Z.; et al. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genom. 2013, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Swanson-Wagner, R.A.; Jia, Y.; DeCook, R.; Borsuk, L.A.; Nettleton, D.; Schnable, P.S. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. USA 2006, 103, 6805–6810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, C.; Fu, F.; Zhong, X.; Peng, B.; Yan, H.; Zhang, J.; Zhang, W.; Wang, P.; Ding, X.; et al. Comparative transcriptome analysis of flower heterosis in two soybean F1 hybrids by RNA-seq. PLoS ONE 2017, 12, e0181061. [Google Scholar] [CrossRef]
- Wang, H.; Fang, Y.; Wang, L.; Zhu, W.; Ji, H.; Wang, H.; Xu, S.; Sima, Y. Heterosis and differential gene expression in hybrids and parents in Bombyx mori by digital gene expression profiling. Sci. Rep. 2015, 5, 8750. [Google Scholar] [CrossRef]
- Cahaner, A.; Smith, E.J.; Swenson, S.; Lamont, S.J. Associations of individual genomic heterozygosity, estimated by molecular fingerprinting, and of dam major histocompatibility complex with growth and egg production traits in layer chickens. Poult. Sci. 1996, 75, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Deeb, N.; Lamont, S.J. Genetic architecture of growth and body composition in unique chicken populations. J. Hered. 2002, 93, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Abasht, B.; Lamont, S.J. Genome-wide association analysis reveals cryptic alleles as an important factor in heterosis for fatness in chicken F2 population. Anim. Genet. 2007, 38, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Nordskog, A.W.; Phillips, R.E. Heterosis in poultry: 5. Reciprocal crosses involving leghorns, heavy breeds and fayoumi. Poult. Sci. 1960, 39, 257–263. [Google Scholar] [CrossRef]
- Saadey, S.M.; Galal, A.; Zaky, H.; El-Dein, A.Z. Diallel crossing analysis for body weight and egg production traits of two native Egyptian and two exotic chicken breeds. Int. J. Poult. Sci. 2008, 7, 64–71. [Google Scholar]
- Zhuo, Z.; Lamont, S.J.; Abasht, B. RNA-seq analyses identify frequent allele specific expression and no evidence of genomic imprinting in specific embryonic tissues of chicken. Sci. Rep. 2017, 7, 11944. [Google Scholar] [CrossRef] [PubMed]
- Washburn, K.W.; Guill, R.A. Relationship of embryo weight as a percent of egg weight to efficiency of feed utilization in the hatched chick. Poult. Sci. 1974, 53, 766–769. [Google Scholar] [CrossRef]
- SAS Insititute Inc. JMP Pro, Version 13.1.0; SAS Insititute Inc.: Cary, NC, USA, 1989–2007. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Olex, A.L.; Fetrow, J.S. SC2ATmd: A tool for integration of the figure of merit with cluster analysis for gene expression data. Bioinformatics 2011, 27, 1330–1331. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Lucas, A. Amap: Another Multidimensional Analysis Package. R package version 0.8-14. Available online: https://rdrr.io/cran/amap/ (accessed on 15 December 2018).
- Huang, D.W.; Sherman, B.T.; Lempicki, R. A systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Takaishi, T.; Ueshima, S.; Matsuo, O. New aspects of fibrinolytic proteins in brain development. Cell Struct. Funct. 1997, 22, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Moonen, G.; Grau-Wagemans, M.P.; Selak, I. Plasminogen activator–plasmin system and neuronal migration. Nature 1982, 298, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Seeds, N.W.; Verrall, S.; Friedman, G.; Hayden, S.; Gadotti, D.; Haffke, S.; Christensen, K.; Gardner, B.; McGUIRE, P.; Krystosek, A. Plasminogen activators and plasminogen activator inhibitors in neural development. Ann. N. Y. Acad. Sci. 1992, 667, 32–40. [Google Scholar] [CrossRef]
- Seeds, N.W.; Basham, M.E.; Haffke, S.P. Neuronal migration is retarded in mice lacking the tissue plasminogen activator gene. Proc. Natl. Acad. Sci. USA 1999, 96, 14118–14123. [Google Scholar] [CrossRef]
- Zappulli, V.; Friis, K.P.; Fitzpatrick, Z.; Maguire, C.A.; Breakefield, X.O. Extracellular vesicles and intercellular communication within the nervous system. J. Clin. Investig. 2016, 126, 1198–1207. [Google Scholar] [CrossRef]
- Zhao, C.; Nguyen, T.; Liu, L.; Sacco, R.E.; Brogden, K.A.; Lehrer, R.I. Gallinacin-3, an inducible epithelial β-defensin in the chicken. Infect. Immun. 2001, 69, 2684–2691. [Google Scholar] [CrossRef]
- Xiao, Y.; Hughes, A.L.; Ando, J.; Matsuda, Y.; Cheng, J.-F.; Skinner-Noble, D.; Zhang, G. A genome-wide screen identifies a single β-defensin gene cluster in the chicken: Implications for the origin and evolution of mammalian defensins. BMC Genom. 2004, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, H.A.; Elazzazy, A.M.; Abuzinadah, O.A.H.; Al-Hejin, A.M.; Mahmoud, M.M.; Harakeh, S.M. Antimicrobial activities of chicken β-defensin (4 and 10) peptides against pathogenic bacteria and fungi. Front. Cell. Infect. Microbiol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.S.; Koltes, J.E.; Fritz-Waters, E.R.; Rothschild, M.F.; Schmidt, C.J.; Ashwell, C.M.; Persia, M.E.; Reecy, J.M.; Lamont, S.J. Single nucleotide variant discovery of highly inbred Leghorn and Fayoumi chicken breeds using pooled whole genome resequencing data reveals insights into phenotype differences. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Dalgaard, T.; Højsgaard, S.; Skjødt, K.; Juul-Madsen, H.R. Differences in chicken major histocompatibility complex (MHC) class Iα gene expression between Marek’s disease-resistant and -susceptible MHC haplotypes. Scand. J. Immunol. 2003, 57, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Chappell, P.E.; Meziane, E.K.; Harrison, M.; Magiera, Ł.; Hermann, C.; Mears, L.; Wrobel, A.G.; Durant, C.; Nielsen, L.L.; Buus, S.; et al. Expression levels of MHC class I molecules are inversely correlated with promiscuity of peptide binding. eLife 2015, 4, e05345. [Google Scholar] [CrossRef]
- Knolle, P.A.; Gerken, G. Local control of the immune response in the liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef]
- Li, L.; Lu, K.; Chen, Z.; Mu, T.; Hu, Z.; Li, X. Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics 2008, 180, 1725–1742. [Google Scholar] [CrossRef]
- Rapp, R.A.; Udall, J.A.; Wendel, J.F. Genomic expression dominance in allopolyploids. BMC Biol. 2009, 7, 18. [Google Scholar] [CrossRef]
- Tomita, K.; Ueda, T.; Watanabe, K. RNA editing in the acceptor stem of squid mitochondrial tRNA(Tyr). Nucleic Acids Res. 1996, 24, 4987–4991. [Google Scholar] [CrossRef]
- Bratic, A.; Clemente, P.; Calvo-Garrido, J.; Maffezzini, C.; Felser, A.; Wibom, R.; Wedell, A.; Freyer, C.; Wredenberg, A. Mitochondrial polyadenylation is a one-step process required for mRNA integrity and tRNA maturation. PLoS Genet. 2016, 12, e1006028. [Google Scholar] [CrossRef]
- Li, Z.; Reimers, S.; Pandit, S.; Deutscher, M.P. RNA quality control: Degradation of defective transfer RNA. EMBO J. 2002, 21, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Kadaba, S.; Krueger, A.; Trice, T.; Krecic, A.M.; Hinnebusch, A.G.; Anderson, J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004, 18, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Vaňáčová, Š.; Wolf, J.; Martin, G.; Blank, D.; Dettwiler, S.; Friedlein, A.; Langen, H.; Keith, G.; Keller, W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005, 3, e189. [Google Scholar] [CrossRef]
- Slomovic, S.; Laufer, D.; Geiger, D.; Schuster, G. Polyadenylation and degradation of human mitochondrial RNA: The prokaryotic past leaves its mark. Mol. Cell. Biol. 2005, 25, 6427–6435. [Google Scholar] [CrossRef] [PubMed]
- Yokobori, S.; Pääbo, S. Polyadenylation creates the discriminator nucleotide of chicken mitochondrial tRNA(Tyr). J. Mol. Biol. 1997, 265, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Kawase, M.; Kawakami, Y.; Ohmori, S. Concentrations of D-lactate and its related metabolic intermediates in liver, blood, and muscle of diabetic and starved rats. Res. Exp. Med. 1992, 192, 407–414. [Google Scholar] [CrossRef]
- Ling, B.; Peng, F.; Alcorn, J.; Lohmann, K.; Bandy, B.; Zello, G.A. D-Lactate altered mitochondrial energy production in rat brain and heart but not liver. Nutr. Metab. 2012, 9, 6. [Google Scholar] [CrossRef]
- Bottje, W.; Kong, B.-W.; Reverter, A.; Waardenberg, A.J.; Lassiter, K.; Hudson, N.J. Progesterone signalling in broiler skeletal muscle is associated with divergent feed efficiency. BMC Syst. Biol. 2017, 11, 29. [Google Scholar] [CrossRef]
- Ullu, E.; Murphy, S.; Melli, M. Human 7SL RNA consists of a 140 nucleotide middle-repetitive sequence inserted in an Alu sequence. Cell 1982, 29, 195–202. [Google Scholar] [CrossRef]
- Liu, W.M.; Dennis, J.L.; Gravett, A.M.; Chanthirakumar, C.; Kaminska, E.; Coulton, G.; Fowler, D.W.; Bodman-Smith, M.; Dalgleish, A.G. Supernatants derived from chemotherapy-treated cancer cell lines can modify angiogenesis. Br. J. Cancer 2012, 106, 896–903. [Google Scholar] [CrossRef]
- Schageman, J.; Zeringer, E.; Li, M.; Barta, T.; Lea, K.; Gu, J.; Magdaleno, S.; Setterquist, R.; Vlassov, A.V. The complete exosome workflow solution: From isolation to characterization of RNA cargo. BioMed Res. Int. 2013, 2013, 253957. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.Y.; Qiu, Y.; Shabason, J.E.; Wu, T.J.; Yoon, T.; Kim, B.C.; Benci, J.L.; DeMichele, A.M.; Tchou, J.; Marcotrigiano, J.; et al. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell 2017, 170, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.B.; Robinson, D.; Hauringa, M.; Raeker, M.O.; Borisov, A.B.; Westfall, M.V.; Russell, M.W. Obscurin-like 1, OBSL1, is a novel cytoskeletal protein related to obscurin. Genomics 2007, 89, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Litterman, N.; Ikeuchi, Y.; Gallardo, G.; O’Connell, B.C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W.; Bonni, A. An OBSL1-Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 2011, 9, e1001060. [Google Scholar] [CrossRef] [PubMed]
- Finlin, B.S.; Andres, D.A. Rem is a new member of the Rad-and Gem/Kir Ras-related GTP-binding protein family repressed by lipopolysaccharide stimulation. J. Biol. Chem. 1997, 272, 21982–21988. [Google Scholar] [CrossRef] [PubMed]
- Ward, Y.; Yap, S.F.; Ravichandran, V.; Matsumura, F.; Ito, M.; Spinelli, B.; Kelly, K. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell Biol. 2002, 157, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Prince, A.M. Quantitative studies on Rous sarcoma virus. II. Mechanism of resistance of chick embryos to chorio-allantoic inoculation of Rous sarcoma virus. J. Natl. Cancer Inst. 1958, 20, 843–850. [Google Scholar]
- Springer, N.M.; Stupar, R.M. Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell 2007, 19, 2391–2402. [Google Scholar] [CrossRef]
- Bell, G.D.M.; Kane, N.C.; Rieseberg, L.H.; Adams, K.L. RNA-seq analysis of allele-specific expression, hybrid effects, and regulatory divergence in hybrids compared with their parents from natural populations. Genome Biol. Evol. 2013, 5, 1309–1323. [Google Scholar] [CrossRef]
- Song, G.; Guo, Z.; Liu, Z.; Cheng, Q.; Qu, X.; Chen, R.; Jiang, D.; Liu, C.; Wang, W.; Sun, Y.; et al. Global RNA sequencing reveals that genotype-dependent allele-specific expression contributes to differential expression in rice F1 hybrids. BMC Plant Biol. 2013, 13, 221. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brain | Liver | |
|---|---|---|
| F vs. L | 304 | 579 |
| F vs. Cross | 310 | 91 |
| L vs. Cross | 73 | 147 |
| FL vs. LF | 5 | 6 |
| MPV vs. Cross | 133 | 40 |
| A | OD | D_F | D_L | ASE_F | ASE_L | |
|---|---|---|---|---|---|---|
| Brain | ||||||
| A | 362 | - | - | - | - | - |
| OD | 0 | 5 | - | - | - | - |
| D_F | 0 | 0 | 13 | - | - | - |
| D_L | 0 | 0 | 0 | 115 | - | - |
| ASE_F | 16 | 0 | 0 | 2 | 674 | - |
| ASE_L | 15 | 0 | 0 | 1 | 0 | 736 |
| Liver | ||||||
| A | 549 | - | - | - | - | - |
| OD | 0 | 2 | - | - | - | - |
| D_F | 0 | 0 | 31 | - | - | - |
| D_L | 0 | 0 | 0 | 7 | - | - |
| ASE_F | 62* | 0 | 0 | 1 | 632 | - |
| ASE_L | 45* | 0 | 0 | 0 | 0 | 608 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuo, Z.; Lamont, S.J.; Abasht, B. RNA-Seq Analyses Identify Additivity as the Predominant Gene Expression Pattern in F1 Chicken Embryonic Brain and Liver. Genes 2019, 10, 27. https://doi.org/10.3390/genes10010027
Zhuo Z, Lamont SJ, Abasht B. RNA-Seq Analyses Identify Additivity as the Predominant Gene Expression Pattern in F1 Chicken Embryonic Brain and Liver. Genes. 2019; 10(1):27. https://doi.org/10.3390/genes10010027
Chicago/Turabian StyleZhuo, Zhu, Susan J. Lamont, and Behnam Abasht. 2019. "RNA-Seq Analyses Identify Additivity as the Predominant Gene Expression Pattern in F1 Chicken Embryonic Brain and Liver" Genes 10, no. 1: 27. https://doi.org/10.3390/genes10010027
APA StyleZhuo, Z., Lamont, S. J., & Abasht, B. (2019). RNA-Seq Analyses Identify Additivity as the Predominant Gene Expression Pattern in F1 Chicken Embryonic Brain and Liver. Genes, 10(1), 27. https://doi.org/10.3390/genes10010027