Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response


Abstract
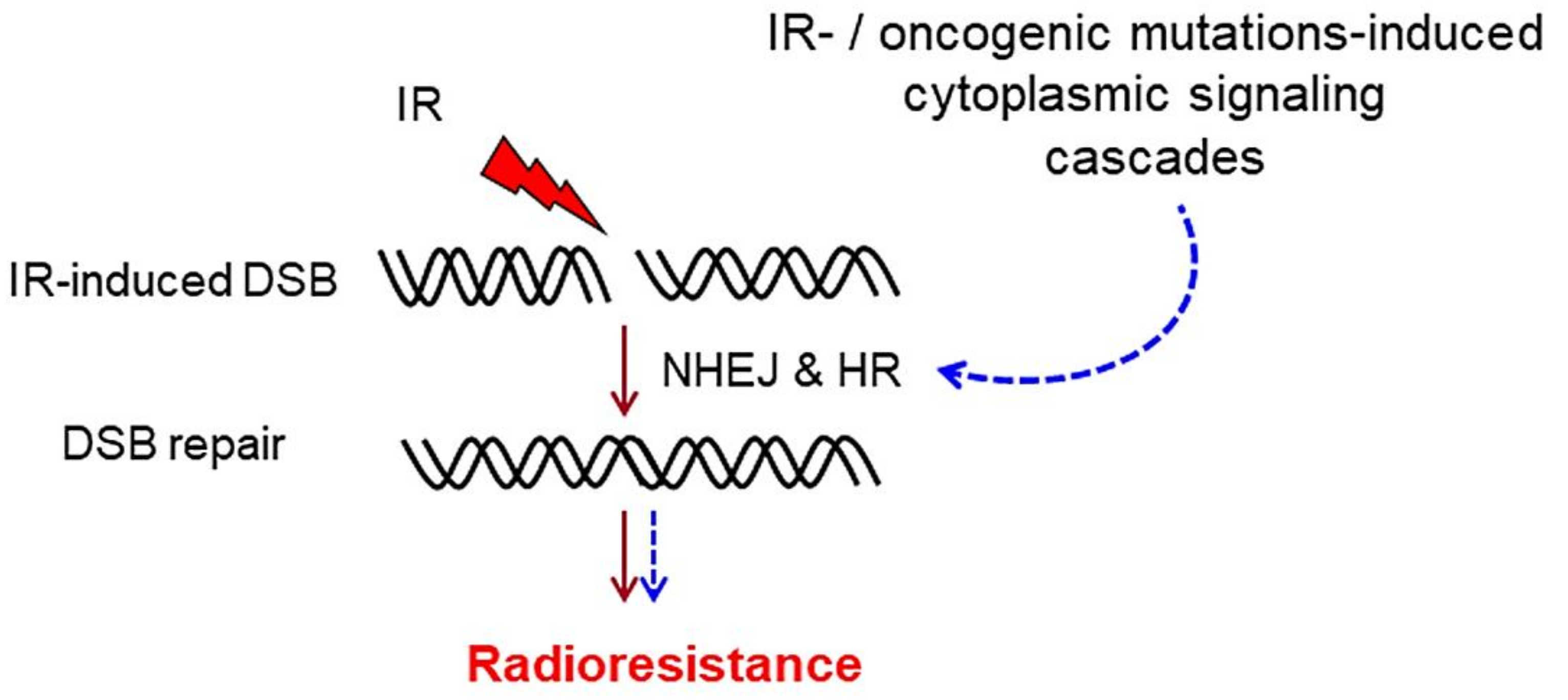
1. DNA Double-Strand Break Repair
2. Receptor Tyrosine Kinases That Mediate DSB Repair after Irradiation
2.1. ErbB Family of RTKs
2.2. Targeting the ErbB Family of RTKs in Combination with Radiotherapy
2.3. IGF-1 Receptor
2.4. Targeting IGF-1R in Combination with Radiotherapy
2.5. TAM Family of Receptors
2.6. Targeting AXL in Combination with Radiotherapy
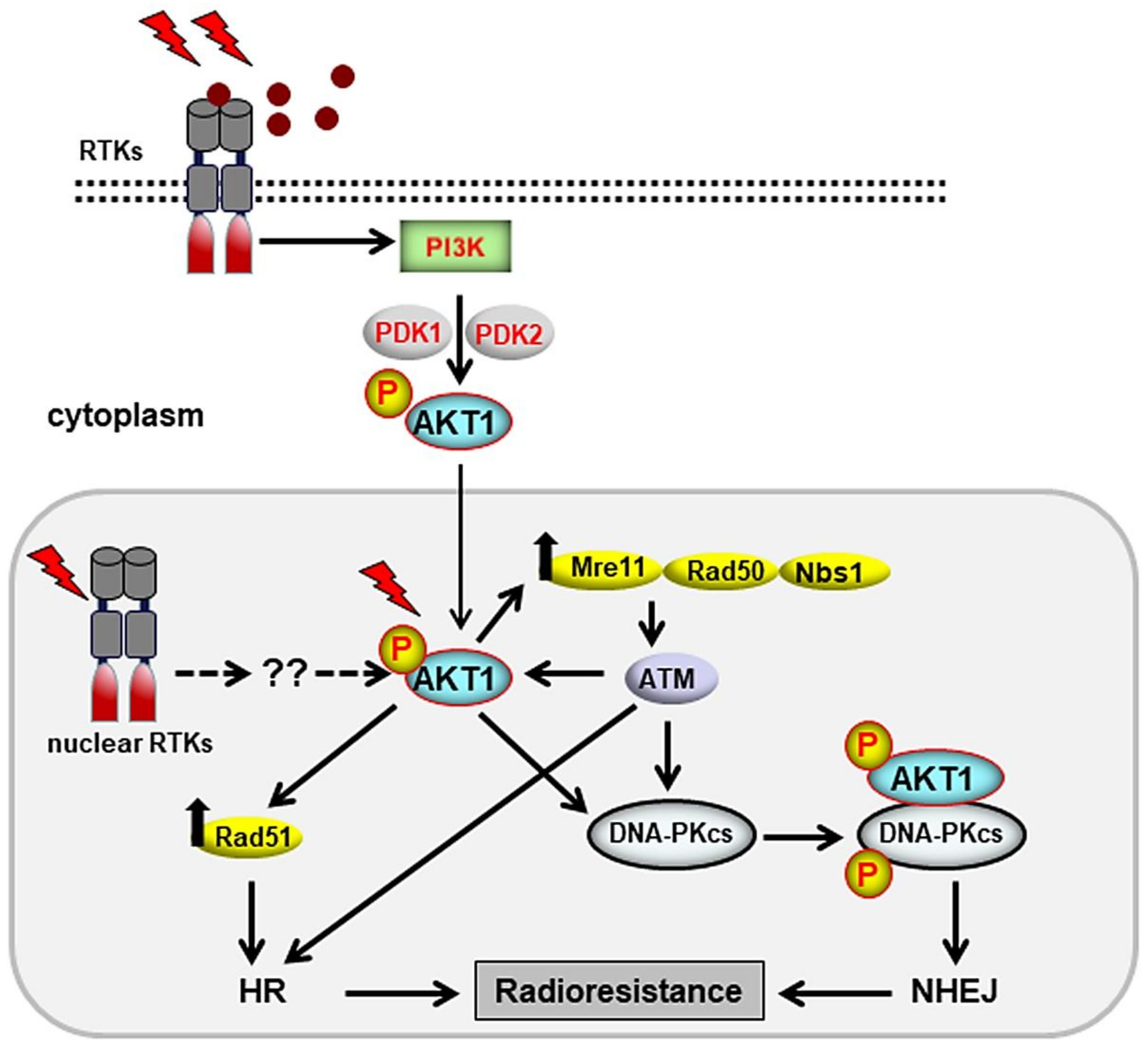
3. Cytoplasmic Signaling Cascades That Stimulate DSB Repair
3.1. Stimulation of DSB Repair by the PI3K/AKT Pathway
3.2. Targeting AKT for Radiosensitization
4. Conclusions and Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Pantelias, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J. Biol. Chem. 2010, 285, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Jones, D.; Lee, S.H.; Williamson, E.A.; Hromas, R. Drugging the cancers addicted to DNA repair. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Williams, R.S.; Williams, J.S.; Tainer, J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 2007, 85, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. Medchemcomm 2017, 8, 295–319. [Google Scholar] [CrossRef]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G. Backup pathways of NHEJ in cells of higher eukaryotes: Cell cycle dependence. Radiother. Oncol. 2009, 92, 310–315. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Geuting, V.; Lobrich, M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. 2011, 101, 7–12. [Google Scholar] [CrossRef]
- Martin, L.M.; Marples, B.; Coffey, M.; Lawler, M.; Lynch, T.H.; Hollywood, D.; Marignol, L. DNA mismatch repair and the DNA damage response to ionizing radiation: Making sense of apparently conflicting data. Cancer Treat. Rev. 2010, 36, 518–527. [Google Scholar] [CrossRef]
- Kennedy, R.D.; D’Andrea, A.D. DNA repair pathways in clinical practice: Lessons from pediatric cancer susceptibility syndromes. J. Clin. Oncol. 2006, 24, 3799–3808. [Google Scholar] [CrossRef] [PubMed]
- Stachelek, G.C.; Peterson-Roth, E.; Liu, Y.; Fernandez, R.J., 3rd; Pike, L.R.; Qian, J.M.; Abriola, L.; Hoyer, D.; Hungerford, W.; Merkel, J.; et al. YU238259 is a novel inhibitor of homology-dependent DNA repair that exhibits synthetic lethality and radiosensitization in repair-deficient tumors. Mol. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Thoms, J.; Bristow, R.G. DNA repair targeting and radiotherapy: A focus on the therapeutic ratio. Semin. Radiat. Oncol. 2010, 20, 217–222. [Google Scholar] [CrossRef]
- Rodemann, H.P.; Dittmann, K.; Toulany, M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int. J. Radiat. Biol. 2007, 83, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Pueyo, G.; Mesia, R.; Figueras, A.; Lozano, A.; Baro, M.; Vazquez, S.; Capella, G.; Balart, J. Cetuximab may inhibit tumor growth and angiogenesis induced by ionizing radiation: A preclinical rationale for maintenance treatment after radiotherapy. Oncologist 2010, 15, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Dittmann, K.; Baumann, M.; Rodemann, H.P. Radiosensitization of Ras-mutated human tumor cells in vitro by the specific EGF receptor antagonist BIBX1382BS. Radiother. Oncol. 2005, 74, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Amorino, G.P.; Hamilton, V.M.; Valerie, K.; Dent, P.; Lammering, G.; Schmidt-Ullrich, R.K. Epidermal growth factor receptor dependence of radiation-induced transcription factor activation in human breast carcinoma cells. Mol. Biol. Cell 2002, 13, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ullrich, R.K.; Mikkelsen, R.B.; Dent, P.; Todd, D.G.; Valerie, K.; Kavanagh, B.D.; Contessa, J.N.; Rorrer, W.K.; Chen, P.B. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene 1997, 15, 1191–1197. [Google Scholar] [CrossRef]
- Contessa, J.N.; Hampton, J.; Lammering, G.; Mikkelsen, R.B.; Dent, P.; Valerie, K.; Schmidt-Ullrich, R.K. Ionizing radiation activates Erb-B receptor dependent AKT and p70 S6 kinase signaling in carcinoma cells. Oncogene 2002, 21, 4032–4041. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Spangle, J.M.; Roberts, T.M. Epigenetic regulation of RTK signaling. J. Mol. Med. 2017, 95, 791–798. [Google Scholar] [CrossRef]
- Song, H.; Li, C.W.; Labaff, A.M.; Lim, S.O.; Li, L.Y.; Kan, S.F.; Chen, Y.; Zhang, K.; Lang, J.; Xie, X.; et al. Acetylation of EGF receptor contributes to tumor cell resistance to histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 2011, 404, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Hung, M.C. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin Cancer Res. 2009, 15, 6484–6489. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K.; Chen, B.P.; Story, M.D.; Sato, M.; Minna, J.D.; Chen, D.J.; Nirodi, C.S. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res. 2007, 67, 5267–5274. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Czemmel, S.; Huber, S.M.; Rodemann, H.P. New roles for nuclear EGFR in regulating the stability and translation of mRNAs associated with VEGF signaling. PLoS ONE 2017, 12, e0189087. [Google Scholar] [CrossRef] [PubMed]
- Faria, J.; de Andrade, C.; Goes, A.M.; Rodrigues, M.A.; Gomes, D.A. Effects of different ligands on epidermal growth factor receptor (EGFR) nuclear translocation. Biochem. Biophys. Res. Commun. 2016, 478, 39–45. [Google Scholar] [CrossRef]
- Minjgee, M.; Toulany, M.; Kehlbach, R.; Giehl, K.; Rodemann, H.P. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent AKT signaling and activation of DNA-PKcs. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1506–1514. [Google Scholar] [CrossRef]
- Saki, M.; Toulany, M.; Rodemann, H.P. Acquired resistance to cetuximab is associated with the overexpression of Ras family members and the loss of radiosensitization in head and neck cancer cells. Radiother. Oncol. 2013, 108, 473–478. [Google Scholar] [CrossRef]
- Toulany, M.; Baumann, M.; Rodemann, H.P. Stimulated PI3K-AKT signaling mediated through ligand or radiation-induced EGFR depends indirectly, but not directly, on constitutive K-Ras activity. Mol. Cancer Res. 2007, 5, 863–872. [Google Scholar] [CrossRef]
- Nowsheen, S.; Cooper, T.; Stanley, J.A.; Yang, E.S. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS ONE 2012, 7, e46614. [Google Scholar] [CrossRef]
- Lee, H.J.; Lan, L.; Peng, G.; Chang, W.C.; Hsu, M.C.; Wang, Y.N.; Cheng, C.C.; Wei, L.; Nakajima, S.; Chang, S.S.; et al. Tyrosine 370 phosphorylation of ATM positively regulates DNA damage response. Cell Res. 2015, 25, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Nakajima, Y.; Yu, Y.L.; Xia, W.; Chen, C.T.; Yang, C.C.; McIntush, E.W.; Li, L.Y.; Hawke, D.H.; Kobayashi, R.; et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell Biol. 2006, 8, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.H.; Wang, Y.N.; Hsieh, Y.H.; Li, L.Y.; Xia, W.; Chang, W.C.; Chang, L.C.; Cheng, C.C.; Lai, C.C.; Hsu, J.L.; et al. EGFR modulates DNA synthesis and repair through Tyr phosphorylation of histone H4. Dev. Cell 2014, 30, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Myllynen, L.; Rieckmann, T.; Dahm-Daphi, J.; Kasten-Pisula, U.; Petersen, C.; Dikomey, E.; Kriegs, M. In tumor cells regulation of DNA double strand break repair through EGF receptor involves both NHEJ and HR and is independent of p53 and K-Ras status. Radiother. Oncol. 2011, 101, 147–151. [Google Scholar] [CrossRef]
- Friedmann, B.J.; Caplin, M.; Savic, B.; Shah, T.; Lord, C.J.; Ashworth, A.; Hartley, J.A.; Hochhauser, D. Interaction of the epidermal growth factor receptor and the DNA-dependent protein kinase pathway following gefitinib treatment. Mol. Cancer Ther. 2006, 5, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Yang, E.S.; Arteaga, C.L.; Xia, F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res. 2008, 68, 9141–9146. [Google Scholar] [CrossRef]
- Li, Y.H.; Wang, X.; Pan, Y.; Lee, D.H.; Chowdhury, D.; Kimmelman, A.C. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS ONE 2012, 7, e39588. [Google Scholar] [CrossRef] [PubMed]
- Milas, L.; Fan, Z.; Andratschke, N.H.; Ang, K.K. Epidermal growth factor receptor and tumor response to radiation: In vivo preclinical studies. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7356. [Google Scholar] [PubMed]
- Iida, M.; Bahrar, H.; Brand, T.M.; Pearson, H.E.; Coan, J.P.; Orbuch, R.A.; Flanigan, B.G.; Swick, A.D.; Prabakaran, P.J.; Lantto, J.; et al. Targeting the HER family with pan-HER effectively overcomes resistance to cetuximab. Mol. Cancer Ther. 2016, 15, 2175–2186. [Google Scholar] [CrossRef]
- Iida, M.; Brand, T.M.; Starr, M.M.; Li, C.; Huppert, E.J.; Luthar, N.; Pedersen, M.W.; Horak, I.D.; Kragh, M.; Wheeler, D.L. Sym004, a novel EGFR antibody mixture, can overcome acquired resistance to cetuximab. Neoplasia 2013, 15, 1196–1206. [Google Scholar] [CrossRef]
- Francis, D.M.; Huang, S.; Armstrong, E.A.; Werner, L.R.; Hullett, C.; Li, C.; Morris, Z.S.; Swick, A.D.; Kragh, M.; Lantto, J.; et al. Pan-HER inhibitor augments radiation response in human lung and head and neck cancer models. Clin. Cancer Res. 2016, 22, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Peter Rodemann, H.; Harari, P.M. Molecular targeting of growth factor receptor signaling in radiation oncology. Recent Results Cancer Res. 2016, 198, 45–87. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Garg, M.K.; Zhao, F.; Sparano, J.A.; Palefsky, J.; Whittington, R.; Mitchell, E.P.; Mulcahy, M.F.; Armstrong, K.I.; Nabbout, N.H.; Kalnicki, S.; et al. Cetuximab plus chemoradiotherapy in immunocompetent patients with anal carcinoma: A phase II Eastern cooperative oncology group-American College of Radiology Imaging Network Cancer Research Group Trial (E3205). J. Clin. Oncol. 2017, 35, 718–726. [Google Scholar] [CrossRef]
- Sparano, J.A.; Lee, J.Y.; Palefsky, J.; Henry, D.H.; Wachsman, W.; Rajdev, L.; Aboulafia, D.; Ratner, L.; Fitzgerald, T.J.; Kachnic, L.; et al. Cetuximab plus chemoradiotherapy for HIV-associated anal carcinoma: A phase II AIDS malignancy consortium trial. J. Clin. Oncol. 2017, 35, 727–733. [Google Scholar] [CrossRef]
- Eisterer, W.; De Vries, A.; Ofner, D.; Rabl, H.; Koplmuller, R.; Greil, R.; Tschmelitsch, J.; Schmid, R.; Kapp, K.; Lukas, P.; et al. Preoperative treatment with capecitabine, cetuximab and radiotherapy for primary locally advanced rectal cancer--a phase II clinical trial. Anticancer Res. 2014, 34, 6767–6773. [Google Scholar]
- Bonomo, P.; Loi, M.; Desideri, I.; Olmetto, E.; Delli Paoli, C.; Terziani, F.; Greto, D.; Mangoni, M.; Scoccianti, S.; Simontacchi, G.; et al. Incidence of skin toxicity in squamous cell carcinoma of the head and neck treated with radiotherapy and cetuximab: A systematic review. Crit. Rev. Oncol. Hematol. 2017, 120, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Lemanski, C.; Pignon, J.P.; Levy, A.; Delarochefordiere, A.; Martel-Lafay, I.; Rio, E.; Malka, D.; Conroy, T.; Miglianico, L.; et al. Unexpected toxicity of cetuximab combined with conventional chemoradiotherapy in patients with locally advanced anal cancer: Results of the UNICANCER ACCORD 16 phase II trial. Ann. Oncol. 2013, 24, 2834–2838. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.D.; Paulus, R.; Komaki, R.; Masters, G.; Blumenschein, G.; Schild, S.; Bogart, J.; Hu, C.; Forster, K.; Magliocco, A.; et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): A randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015, 16, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Crosby, T.; Hurt, C.N.; Falk, S.; Gollins, S.; Mukherjee, S.; Staffurth, J.; Ray, R.; Bashir, N.; Bridgewater, J.A.; Geh, J.I.; et al. Chemoradiotherapy with or without cetuximab in patients with oesophageal cancer (SCOPE1): A multicentre, phase 2/3 randomised trial. Lancet Oncol. 2013, 14, 627–637. [Google Scholar] [CrossRef]
- Ang, K.K.; Zhang, Q.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Sherman, E.J.; Weber, R.S.; Galvin, J.M.; Bonner, J.A.; Harris, J.; El-Naggar, A.K.; et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J. Clin. Oncol. 2014, 32, 2940–2950. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Trotti, A.M.; Harris, J.; Eisbruch, A.; Harari, P.M.; Adelstein, D.J.; Sturgis, E.M.; Burtness, B.; Ridge, J.A.; Ringash, J.; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet 2018. [Google Scholar] [CrossRef]
- Chang, C.C.; Chi, K.H.; Kao, S.J.; Hsu, P.S.; Tsang, Y.W.; Chang, H.J.; Yeh, Y.W.; Hsieh, Y.S.; Jiang, J.S. Upfront gefitinib/erlotinib treatment followed by concomitant radiotherapy for advanced lung cancer: A mono-institutional experience. Lung Cancer 2011, 73, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xia, T.Y.; Wang, Y.J.; Li, H.Q.; Li, P.; Wang, J.D.; Chang, D.S.; Liu, L.Y.; Di, Y.P.; Wang, X.; et al. Prospective study of epidermal growth factor receptor tyrosine kinase inhibitors concurrent with individualized radiotherapy for patients with locally advanced or metastatic non-small-cell lung cancer. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, e59–e65. [Google Scholar] [CrossRef]
- Iyengar, P.; Kavanagh, B.D.; Wardak, Z.; Smith, I.; Ahn, C.; Gerber, D.E.; Dowell, J.; Hughes, R.; Abdulrahman, R.; Camidge, D.R.; et al. Phase II trial of stereotactic body radiation therapy combined with erlotinib for patients with limited but progressive metastatic non-small-cell lung cancer. J. Clin. Oncol. 2014, 32, 3824–3830. [Google Scholar] [CrossRef]
- Casal Rubio, J.; Firvida-Perez, J.L.; Lazaro-Quintela, M.; Baron-Duarte, F.J.; Alonso-Jaudenes, G.; Santome, L.; Afonso-Afonso, F.J.; Amenedo, M.; Huidobro, G.; Campos-Balea, B.; et al. A phase II trial of erlotinib as maintenance treatment after concurrent chemoradiotherapy in stage III non-small-cell lung cancer (NSCLC): A Galician Lung Cancer Group (GGCP) study. Cancer Chemother. Pharmacol. 2014, 73, 451–457. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Spigel, D.R.; Greco, F.A.; Shipley, D.L.; Peyton, J.; Rubin, M.; Stipanov, M.; Meluch, A. Combined modality treatment with chemotherapy, radiation therapy, bevacizumab, and erlotinib in patients with locally advanced squamous carcinoma of the head and neck: A phase II trial of the Sarah Cannon oncology research consortium. Cancer J. 2011, 17, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.L.; Molinaro, A.M.; Phillips, J.J.; Butowski, N.A.; Chang, S.M.; Perry, A.; Costello, J.F.; DeSilva, A.A.; Rabbitt, J.E.; Prados, M.D. A single-institution phase II trial of radiation, temozolomide, erlotinib, and bevacizumab for initial treatment of glioblastoma. Neuro-Oncology 2014, 16, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Harari, P.M.; Huang, S.M. Head and neck cancer as a clinical model for molecular targeting of therapy: Combining EGFR blockade with radiation. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 427–433. [Google Scholar] [CrossRef]
- Xie, Y.; Hung, M.C. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochem. Biophys. Res. Commun. 1994, 203, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Lien, H.C.; Xia, W.; Chen, I.F.; Lo, H.W.; Wang, Z.; Ali-Seyed, M.; Lee, D.F.; Bartholomeusz, G.; Ou-Yang, F.; et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 2004, 6, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Reif, R.; Adawy, A.; Vartak, N.; Schroder, J.; Gunther, G.; Ghallab, A.; Schmidt, M.; Schormann, W.; Hengstler, J.G. Activated ErbB3 translocates to the nucleus via clathrin-independent endocytosis, which is associated with proliferating cells. J. Biol. Chem. 2016, 291, 3837–3847. [Google Scholar] [CrossRef]
- Huang, S.; Li, C.; Armstrong, E.A.; Peet, C.R.; Saker, J.; Amler, L.C.; Sliwkowski, M.X.; Harari, P.M. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013, 73, 824–833. [Google Scholar] [CrossRef]
- Hagerstrand, D.; Lindh, M.B.; Pena, C.; Garcia-Echeverria, C.; Nister, M.; Hofmann, F.; Ostman, A. PI3K/PTEN/AKT pathway status affects the sensitivity of high-grade glioma cell cultures to the insulin-like growth factor-1 receptor inhibitor NVP-AEW541. Neuro-Oncology 2010, 12, 967–975. [Google Scholar] [CrossRef]
- Waraky, A.; Lin, Y.; Warsito, D.; Haglund, F.; Aleem, E.; Larsson, O. Nuclear insulin-like growth factor 1 receptor phosphorylates proliferating cell nuclear antigen and rescues stalled replication forks after DNA damage. J. Biol. Chem. 2017, 292, 18227–18239. [Google Scholar] [CrossRef]
- Codony-Servat, J.; Cuatrecasas, M.; Asensio, E.; Montironi, C.; Martinez-Cardus, A.; Marin-Aguilera, M.; Horndler, C.; Martinez-Balibrea, E.; Rubini, M.; Jares, P.; et al. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br. J. Cancer 2017, 117, 1777–1786. [Google Scholar] [CrossRef]
- Aleksic, T.; Verrill, C.; Bryant, R.J.; Han, C.; Worrall, A.R.; Brureau, L.; Larre, S.; Higgins, G.S.; Fazal, F.; Sabbagh, A.; et al. IGF-1R associates with adverse outcomes after radical radiotherapy for prostate cancer. Br. J. Cancer 2017, 117, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Lodhia, K.A.; Gao, S.; Aleksic, T.; Esashi, F.; Macaulay, V.M. Suppression of homologous recombination sensitizes human tumor cells to IGF-1R inhibition. Int. J. Cancer 2015, 136, 2961–2966. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, M.M.; Lodhia, K.A.; Aleksic, T.; Gao, S.; Protheroe, A.S.; Macaulay, V.M. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene 2014, 33, 5262–5273. [Google Scholar] [CrossRef] [PubMed]
- Turney, B.W.; Kerr, M.; Chitnis, M.M.; Lodhia, K.; Wang, Y.; Riedemann, J.; Rochester, M.; Protheroe, A.S.; Brewster, S.F.; Macaulay, V.M. Depletion of the type 1 IGF receptor delays repair of radiation-induced DNA double strand breaks. Radiother. Oncol. 2012, 103, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.W.; Saba, C.; Armstrong, E.A.; Huang, S.M.; Benavente, S.; Ludwig, D.L.; Hicklin, D.J.; Harari, P.M. Insulin-like growth factor-I receptor signaling blockade combined with radiation. Cancer Res. 2007, 67, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, T.; Okamoto, I.; Suzuki, M.; Hatashita, E.; Yamada, Y.; Fukuoka, M.; Ono, K.; Nakagawa, K. Inhibition of insulin-like growth factor 1 receptor by CP-751,871 radiosensitizes non-small cell lung cancer cells. Clin. Cancer Res. 2009, 15, 5117–5125. [Google Scholar] [CrossRef] [PubMed]
- Isebaert, S.F.; Swinnen, J.V.; McBride, W.H.; Haustermans, K.M. Insulin-like growth factor-type 1 receptor inhibitor NVP-AEW541 enhances radiosensitivity of PTEN wild-type but not PTEN-deficient human prostate cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 239–247. [Google Scholar] [CrossRef]
- Zhao, H.; Gu, X. Silencing of insulin-like growth factor-1 receptor enhances the radiation sensitivity of human esophageal squamous cell carcinoma in vitro and in vivo. World J. Surg. Oncol. 2014, 12, 325. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Z.; Tang, H.; Jiang, Z.; You, L.; Liao, Y. Crosstalk between IGF-1R and other tumor promoting pathways. Curr. Pharm. Des. 2014, 20, 2912–2921. [Google Scholar] [CrossRef]
- Matsubara, J.; Yamada, Y.; Nakajima, T.E.; Kato, K.; Hamaguchi, T.; Shirao, K.; Shimada, Y.; Shimoda, T. Clinical significance of insulin-like growth factor type 1 receptor and epidermal growth factor receptor in patients with advanced gastric cancer. Oncology 2008, 74, 76–83. [Google Scholar] [CrossRef]
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hulse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK mediates intrinsic and adaptive resistance to AXL-targeting agents. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef]
- Kasikara, C.; Kumar, S.; Kimani, S.; Tsou, W.I.; Geng, K.; Davra, V.; Sriram, G.; Devoe, C.; Nguyen, K.N.; Antes, A.; et al. Phosphatidylserine sensing by TAM receptors regulates AKT-dependent chemoresistance and PD-L1 expression. Mol. Cancer Res. 2017, 15, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Z.; Wang, Z.J.; De, W.; Zheng, M.; Xu, W.Z.; Wu, H.F.; Armstrong, A.; Zhu, J.G. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 2017, 8, 41064–41077. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajeshkumar, N.V.; Maitra, A.; et al. Small-molecule inhibition of Axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Balaji, K.; Vijayaraghavan, S.; Diao, L.; Tong, P.; Fan, Y.; Carey, J.P.; Bui, T.N.; Warner, S.; Heymach, J.V.; Hunt, K.K.; et al. AXL inhibition suppresses the DNA damage response and sensitizes cells to PARP inhibition in multiple cancers. Mol. Cancer Res. 2017, 15, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Skinner, H.D.; Giri, U.; Yang, L.P.; Kumar, M.; Liu, Y.; Story, M.D.; Pickering, C.R.; Byers, L.A.; Williams, M.D.; Wang, J.; et al. Integrative analysis identifies a novel AXL-PI3 Kinase-PD-L1 signaling axis associated with radiation resistance in head and neck cancer. Clin. Cancer Res. 2017, 23, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.; Nelson, M.; Secreto, C.; Warner, S.L.; Bearss, D.J.; Lesnick, C.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. Targeted Axl inhibition primes chronic lymphocytic leukemia B cells to apoptosis and shows synergistic/additive effects in combination with BTK inhibitors. Clin. Cancer Res. 2015, 21, 2115–2126. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Aveic, S.; Corallo, D.; Porcu, E.; Pantile, M.; Boso, D.; Zanon, C.; Viola, G.; Sidarovich, V.; Mariotto, E.; Quattrone, A.; et al. TP-0903 inhibits neuroblastoma cell growth and enhances the sensitivity to conventional chemotherapy. Eur. J. Pharmacol. 2018, 818, 435–448. [Google Scholar] [CrossRef]
- Zhen, Y.; Lee, I.J.; Finkelman, F.D.; Shao, W.H. Targeted inhibition of Axl receptor tyrosine kinase ameliorates anti-GBM-induced lupus-like nephritis. J. Autoimmun. 2018, 93, 37–44. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Pearson, H.E.; Bahrar, H.; Fowler, T.L.; Bednarz, B.P.; et al. AXL is a logical molecular target in head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Flanigan, B.G.; Stein, A.P.; Salgia, R.; Rolff, J.; Kimple, R.J.; et al. The receptor tyrosine kinase AXL mediates nuclear translocation of the epidermal growth factor receptor. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Roberts, T.M. Human tumor mutants in the p110α subunit of PI3K. Cell Cycle 2006, 5, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Zhang, T.; Cui, G.B.; Zhang, J.; Zhang, F.; Zhou, Y.A.; Jiang, T.; Li, X.F. Inhibition of PI3 kinases enhances the sensitivity of non-small cell lung cancer cells to ionizing radiation. Oncol. Rep. 2010, 24, 1683–1689. [Google Scholar] [PubMed]
- Valerie, K.; Yacoub, A.; Hagan, M.P.; Curiel, D.T.; Fisher, P.B.; Grant, S.; Dent, P. Radiation-induced cell signaling: Inside-out and outside-in. Mol. Cancer Ther. 2007, 6, 789–801. [Google Scholar] [CrossRef]
- Toulany, M.; Minjgee, M.; Kehlbach, R.; Chen, J.; Baumann, M.; Rodemann, H.P. ErbB2 expression through heterodimerization with erbB1 is necessary for ionizing radiation- but not EGF-induced activation of AKT survival pathway. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2010, 97, 338–345. [Google Scholar] [CrossRef]
- Li, H.F.; Kim, J.S.; Waldman, T. Radiation-induced AKT activation modulates radioresistance in human glioblastoma cells. Radiat. Oncol. 2009, 4, 43. [Google Scholar] [CrossRef]
- Winograd-Katz, S.E.; Levitzki, A. Cisplatin induces PKB/AKT activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene 2006, 25, 7381–7390. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.C.; Hung, S.K.; Lin, H.Y.; Chiou, W.Y.; Lee, M.S.; Liao, H.F.; Huang, H.B.; Ho, H.C.; Su, Y.C. Targeting the PI3K/AKT/mTOR signaling pathway as an effectively radiosensitizing strategy for treating human oral squamous cell carcinoma in vitro and in vivo. Oncotarget 2017, 8, 68641–68653. [Google Scholar] [CrossRef] [PubMed]
- Bussink, J.; van der Kogel, A.J.; Kaanders, J.H. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol. 2008, 9, 288–296. [Google Scholar] [CrossRef]
- Toulany, M.; Dittmann, K.; Kruger, M.; Baumann, M.; Rodemann, H.P. Radioresistance of K-Ras mutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKT pathway. Radiother. Oncol. 2005, 76, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Han, Z.C. Phosphatidylinositide 3-kinase/AKT in radiation responses. Histol. Histopathol. 2004, 19, 915–923. [Google Scholar] [PubMed]
- Liu, Y.; Cui, B.; Qiao, Y.; Zhang, Y.; Tian, Y.; Jiang, J.; Ma, D.; Kong, B. Phosphoinositide-3-kinase inhibition enhances radiosensitization of cervical cancer in vivo. Int. J. Gynecol. Cancer 2011, 21, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/AKT signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef]
- Choi, E.J.; Ryu, Y.K.; Kim, S.Y.; Wu, H.G.; Kim, J.S.; Kim, I.H.; Kim, I.A. Targeting epidermal growth factor receptor-associated signaling pathways in non-small cell lung cancer cells: Implication in radiation response. Mol. Cancer Res. 2010, 8, 1027–1036. [Google Scholar] [CrossRef]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef]
- Greenwell, I.B.; Ip, A.; Cohen, J.B. PI3K inhibitors: Understanding toxicity mechanisms and management. Oncology 2017, 31, 821–828. [Google Scholar]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/AKT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Xue, G.; Parcellier, A.; Bozulic, L.; Hemmings, B.A. Protein kinase B (PKB/AKT), a key mediator of the PI3K signaling pathway. Curr. Top. Microbiol. Immunol. 2010, 346, 31–56. [Google Scholar] [CrossRef]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef]
- Golding, S.E.; Morgan, R.N.; Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther. 2009, 8, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Harding, S.M.; Zhao, H.; Coackley, C.; Durocher, D.; Bristow, R.G. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 2011, 10, 2218–2232. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. AKT promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Mueck, K.; Rebholz, S.; Harati, M.D.; Rodemann, H.P.; Toulany, M. AKT1 stimulates homologous recombination repair of DNA double-strand breaks in a Rad51-dependent manner. Int. J. Mol. Sci. 2017, 18, 2473. [Google Scholar] [CrossRef]
- Gupta, A.K.; McKenna, W.G.; Weber, C.N.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Machtay, M.; Rosenthal, D.I.; Bakanauskas, V.J.; Cerniglia, G.J.; et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clin. Cancer Res. 2002, 8, 885–892. [Google Scholar]
- Kim, T.J.; Lee, J.W.; Song, S.Y.; Choi, J.J.; Choi, C.H.; Kim, B.G.; Lee, J.H.; Bae, D.S. Increased expression of pAKT is associated with radiation resistance in cervical cancer. Br. J. Cancer 2006, 94, 1678–1682. [Google Scholar] [CrossRef]
- Toulany, M.; Minjgee, M.; Saki, M.; Holler, M.; Meier, F.; Eicheler, W.; Rodemann, H.P. ERK2-dependent reactivation of AKT mediates the limited response of tumor cells with constitutive K-RAS activity to PI3K inhibition. Cancer Biol. Ther. 2014, 15, 317–328. [Google Scholar] [CrossRef]
- Toulany, M.; Iida, M.; Keinath, S.; Iyi, F.F.; Mueck, K.; Fehrenbacher, B.; Mansour, W.Y.; Schaller, M.; Wheeler, D.L.; Rodemann, H.P. Dual targeting of PI3K and MEK enhances the radiation response of K-RAS mutated non-small cell lung cancer. Oncotarget 2016, 7, 43746–43761. [Google Scholar] [CrossRef]
- Zhang, J.; Park, D.; Shin, D.M.; Deng, X. Targeting KRAS-mutant non-small cell lung cancer: Challenges and opportunities. Acta Biochim. Biophys. Sin. 2016, 48, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Kuger, S.; Graus, D.; Brendtke, R.; Gunther, N.; Katzer, A.; Lutyj, P.; Polat, B.; Chatterjee, M.; Sukhorukov, V.L.; Flentje, M.; et al. Radiosensitization of glioblastoma cell lines by the dual PI3K and mTOR inhibitor NVP-BEZ235 depends on drug-irradiation schedule. Transl. Oncol. 2013, 6, 169–179. [Google Scholar] [CrossRef]
- Sathe, A.; Chalaud, G.; Oppolzer, I.; Wong, K.Y.; von Busch, M.; Schmid, S.C.; Tong, Z.; Retz, M.; Gschwend, J.E.; Schulz, W.A.; et al. Parallel PI3K, AKT and mTOR inhibition is required to control feedback loops that limit tumor therapy. PLoS ONE 2018, 13, e0190854. [Google Scholar] [CrossRef] [PubMed]
- Clement, E.; Inuzuka, H.; Nihira, N.T.; Wei, W.; Toker, A. Skp2-dependent reactivation of AKT drives resistance to PI3K inhibitors. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.F.; Defeo-Jones, D.; Fu, S.; Hancock, P.J.; Haskell, K.M.; Jones, R.E.; Kahana, J.A.; Kral, A.M.; Leander, K.; Lee, L.L.; et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific AKT inhibitors. Biochem. J. 2005, 385, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Masure, S.; Haefner, B.; Wesselink, J.J.; Hoefnagel, E.; Mortier, E.; Verhasselt, P.; Tuytelaars, A.; Gordon, R.; Richardson, A. Molecular cloning, expression and characterization of the human serine/threonine kinase AKT-3. Eur. J. Biochem. 1999, 265, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Feng, J.; Li, Y.; Hammarsten, O.; Brazil, D.P.; Hemmings, B.A. DNA-dependent protein kinase-mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C-terminal hydrophobic motif. J. Biol. Chem. 2009, 284, 6169–6174. [Google Scholar] [CrossRef]
- Toulany, M.; Maier, J.; Iida, M.; Rebholz, S.; Holler, M.; Grottke, A.; Juker, M.; Wheeler, D.L.; Rothbauer, U.; Rodemann, H.P. AKT1 and AKT3 but not AKT2 through interaction with DNA-PKcs stimulate proliferation and post-irradiation cell survival of K-RAS-mutated cancer cells. Cell. Death Discov. 2017, 3, 17072. [Google Scholar] [CrossRef] [PubMed]
- Povirk, L.F.; Zhou, R.Z.; Ramsden, D.A.; Lees-Miller, S.P.; Valerie, K. Phosphorylation in the serine/threonine 2609-2647 cluster promotes but is not essential for DNA-dependent protein kinase-mediated nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res. 2007, 35, 3869–3878. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Lobrich, M.; Shiloh, Y.; Chen, D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Schickfluss, T.A.; Fattah, K.R.; Lee, K.J.; Chen, B.P.; Fehrenbacher, B.; Schaller, M.; Chen, D.J.; Rodemann, H.P. Function of erbB receptors and DNA-PKcs on phosphorylation of cytoplasmic and nuclear Akt at S473 induced by erbB1 ligand and ionizing radiation. Radiother. Oncol. 2011, 101, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBalpha/AKT1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Szymonowicz, K.; Oeck, S.; Krysztofiak, A.; van der Linden, J.; Iliakis, G.; Jendrossek, V. Restraining AKT1 phosphorylation attenuates the repair of radiation-induced DNA double-strand breaks and reduces the survival of irradiated cancer cells. Int. J. Mol. Sci. 2018, 19, 2233. [Google Scholar] [CrossRef]
- Holler, M.; Grottke, A.; Mueck, K.; Manes, J.; Jucker, M.; Rodemann, H.P.; Toulany, M. Dual targeting of Akt and mTORC1 impairs repair of dna double-strand breaks and increases radiation sensitivity of human tumor cells. PLoS ONE 2016, 11, e0154745. [Google Scholar] [CrossRef]
- Toulany, M.; Rodemann, H.P. Potential of Akt mediated DNA repair in radioresistance of solid tumors overexpressing erbB-PI3K-AKT pathway. Transl. Cancer Res. 2013, 3, 190–202. [Google Scholar] [CrossRef]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin. Cancer Biol. 2015. [Google Scholar] [CrossRef]
- Philip, C.A.; Laskov, I.; Beauchamp, M.C.; Marques, M.; Amin, O.; Bitharas, J.; Kessous, R.; Kogan, L.; Baloch, T.; Gotlieb, W.H.; et al. Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 2017, 17, 638. [Google Scholar] [CrossRef]
- Deng, R.; Tang, J.; Ma, J.G.; Chen, S.P.; Xia, L.P.; Zhou, W.J.; Li, D.D.; Feng, G.K.; Zeng, Y.X.; Zhu, X.F. PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene 2011, 30, 944–955. [Google Scholar] [CrossRef]
- Viniegra, J.G.; Martinez, N.; Modirassari, P.; Hernandez Losa, J.; Parada Cobo, C.; Sanchez-Arevalo Lobo, V.J.; Aceves Luquero, C.I.; Alvarez-Vallina, L.; Ramon y Cajal, S.; Rojas, J.M.; et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J. Biol. Chem. 2005, 280, 4029–4036. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Khalil, A.; McEwen, A.; Holmes, M.; Neill, S.; Povirk, L.F.; Valerie, K. Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J. Biol. Chem. 2004, 279, 15402–15410. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Kim, J.H.; Paeng, J.Y.; Kim, M.J.; Hong, S.D.; Lee, J.I.; Hong, S.P. Prognostic value of activated AKT expression in oral squamous cell carcinoma. J. Clin. Pathol. 2005, 58, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Ringel, M.D.; Hayre, N.; Saito, J.; Saunier, B.; Schuppert, F.; Burch, H.; Bernet, V.; Burman, K.D.; Kohn, L.D.; Saji, M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001, 61, 6105–6111. [Google Scholar]
- Dobashi, Y.; Kimura, M.; Matsubara, H.; Endo, S.; Inazawa, J.; Ooi, A. Molecular alterations in AKT and its protein activation in human lung carcinomas. Hum. Pathol. 2012, 43, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Tanno, S.; De Rienzo, A.; Klein-Szanto, A.J.; Skele, K.L.; Hoffman, J.P.; Testa, J.R. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J. Cell Biochem. 2002, 87, 470–476. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.G.; Muschel, R.J.; Gupta, A.K.; Hahn, S.M.; Bernhard, E.J. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene 2003, 22, 5866–5875. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Cohen, Y.; Shalmon, B.; Korach, J.; Barshack, I.; Fridman, E.; Rechavi, G. AKT1 pleckstrin homology domain E17K activating mutation in endometrial carcinoma. Gynecol. Oncol. 2010, 116, 88–91. [Google Scholar] [CrossRef]
- Askham, J.M.; Platt, F.; Chambers, P.A.; Snowden, H.; Taylor, C.F.; Knowles, M.A. AKT1 mutations in bladder cancer: Identification of a novel oncogenic mutation that can co-operate with E17K. Oncogene 2010, 29, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Malanga, D.; Scrima, M.; De Marco, C.; Fabiani, F.; De Rosa, N.; De Gisi, S.; Malara, N.; Savino, R.; Rocco, G.; Chiappetta, G.; et al. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle 2008, 7, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1(E17K) in human solid tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Al-Refae, K.; Riffkin, H.; Wiel, G.; Handrick, R.; Klein, D.; Iliakis, G.; Jendrossek, V. Activating AKT1 mutations alter DNA double strand break repair and radiosensitivity. Sci. Rep. 2017, 7, 42700. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.S.; Fedrigo, C.A.; Brands, E.; Dik, R.; Stalpers, L.J.; Baumert, B.G.; Slotman, B.J.; Westerman, B.A.; Peters, G.J.; Sminia, P. The allosteric AKT inhibitor MK2206 shows a synergistic interaction with chemotherapy and radiotherapy in glioblastoma spheroid cultures. BMC Cancer 2017, 17, 204. [Google Scholar] [CrossRef]
- Vink, S.R.; Lagerwerf, S.; Mesman, E.; Schellens, J.H.; Begg, A.C.; van Blitterswijk, W.J.; Verheij, M. Radiosensitization of squamous cell carcinoma by the alkylphospholipid perifosine in cell culture and xenografts. Clin. Cancer Res. 2006, 12, 1615–1622. [Google Scholar] [CrossRef]
- Vink, S.R.; Schellens, J.H.; Beijnen, J.H.; Sindermann, H.; Engel, J.; Dubbelman, R.; Moppi, G.; Hillebrand, M.J.; Bartelink, H.; Verheij, M. Phase I and pharmacokinetic study of combined treatment with perifosine and radiation in patients with advanced solid tumours. Radiother. Oncol. 2006, 80, 207–213. [Google Scholar] [CrossRef]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; McKenna, W.G.; Muschel, R.J. HIV protease inhibitors block AKT signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005, 65, 8256–8265. [Google Scholar] [CrossRef]
- Wilson, J.M.; Fokas, E.; Dutton, S.J.; Patel, N.; Hawkins, M.A.; Eccles, C.; Chu, K.Y.; Durrant, L.; Abraham, A.G.; Partridge, M.; et al. ARCII: A phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer. Radiother. Oncol. 2016, 119, 306–311. [Google Scholar] [CrossRef]
- Hill, E.J.; Roberts, C.; Franklin, J.M.; Enescu, M.; West, N.; MacGregor, T.P.; Chu, K.Y.; Boyle, L.; Blesing, C.; Wang, L.M.; et al. Clinical trial of oral nelfinavir before and during radiation therapy for advanced rectal cancer. Clin. Cancer Res. 2016, 22, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, J.; Lammering, G.; Jansen, R.L.; Beets, G.L.; Wals, J.; Sosef, M.; Den Boer, M.O.; Leijtens, J.; Riedl, R.G.; Theys, J.; et al. Phase I trial of the combination of the Akt inhibitor nelfinavir and chemoradiation for locally advanced rectal cancer. Radiother. Oncol. 2013, 107, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Rengan, R.; Mick, R.; Pryma, D.; Rosen, M.A.; Lin, L.L.; Maity, A.M.; Evans, T.L.; Stevenson, J.P.; Langer, C.J.; Kucharczuk, J.; et al. A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: A report of toxicities and clinical response. J. Thorac. Oncol. 2012, 7, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Brunner, T.B.; Geiger, M.; Grabenbauer, G.G.; Lang-Welzenbach, M.; Mantoni, T.S.; Cavallaro, A.; Sauer, R.; Hohenberger, W.; McKenna, W.G. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J. Clin. Oncol. 2008, 26, 2699–2706. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Target/Drug | Combination | Tumor Type | Outcome | Reference |
|---|---|---|---|---|
| EGFR/Cetuximab | RT | HNSCC | Improved OS | [46,47] |
| CRT | NSCLC/Stage III | No improved OS | [53] | |
| CRT | Esophageal carcinoma | Reduced OS | [54] | |
| CRT | HNSCC | No improved OS | [55] | |
| RT vs. CRT | HPV-positive oropharyngeal carcinoma | Lower PFS after cetuximab + RT compared to CRT | [56] | |
| EGFR/Erlotinib | RT | NSCLC | OS 62.5% (3 years) | [57] |
| RT | Advanced or metastatic NSCLC | OS 30% (3 years) | [58] | |
| SBRT | NSCLC | PFS and OS greater than historical values | [59] | |
| CRT | NSCLC | Effective maintenance therapy PFS 63.5% | [60] | |
| Bavacizumab + CRT | HNSCC | OS 71% and PFS 82% (3 years) | [61] | |
| CRT | GM | No improvement in OS and PFS | [62] |
| Target/Drug | Combination | Tumor Type | Outcome | Reference |
|---|---|---|---|---|
| AKT/Nelfinavir | CRT/Phase II | Pancreatic cancer | Acceptable toxicity and promising activity | [160] |
| RT/Phase I | Rectal cancer | Well-tolerated and good tumor regression | [161] | |
| CRT/Phase I | Rectal cancer | Nelfinavir 750 mg recommended phase II | [162] | |
| CRT/Phase I | NSCLC | Acceptable toxicity and promising activity | [163] | |
| CRT/Phase I | Pancreatic cancer | Acceptable toxicity and promising activity | [164] | |
| AKT/Perifosine | RT/Phase I | NSCLC, prostate, esophageal, colon, and bladder cancer | Recommended phase II, 150 mg/day, started one week prior to RT | [158] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. https://doi.org/10.3390/genes10010025
Toulany M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes. 2019; 10(1):25. https://doi.org/10.3390/genes10010025
Chicago/Turabian StyleToulany, Mahmoud. 2019. "Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response" Genes 10, no. 1: 25. https://doi.org/10.3390/genes10010025
APA StyleToulany, M. (2019). Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes, 10(1), 25. https://doi.org/10.3390/genes10010025