Focus on Hypoxia-Related Pathways in Pediatric Osteosarcomas and Their Druggability

 , ,
, , 
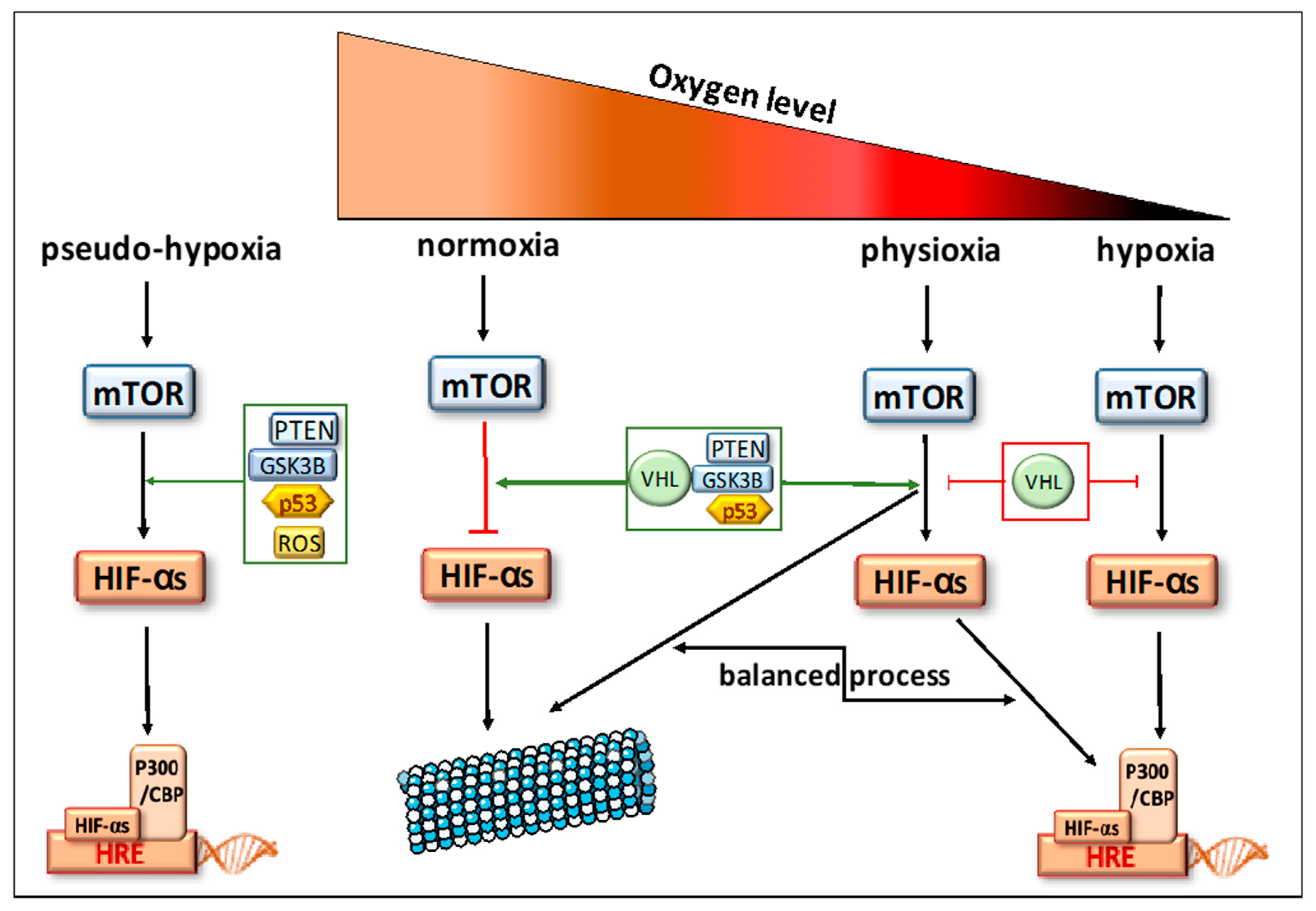{kind=link}
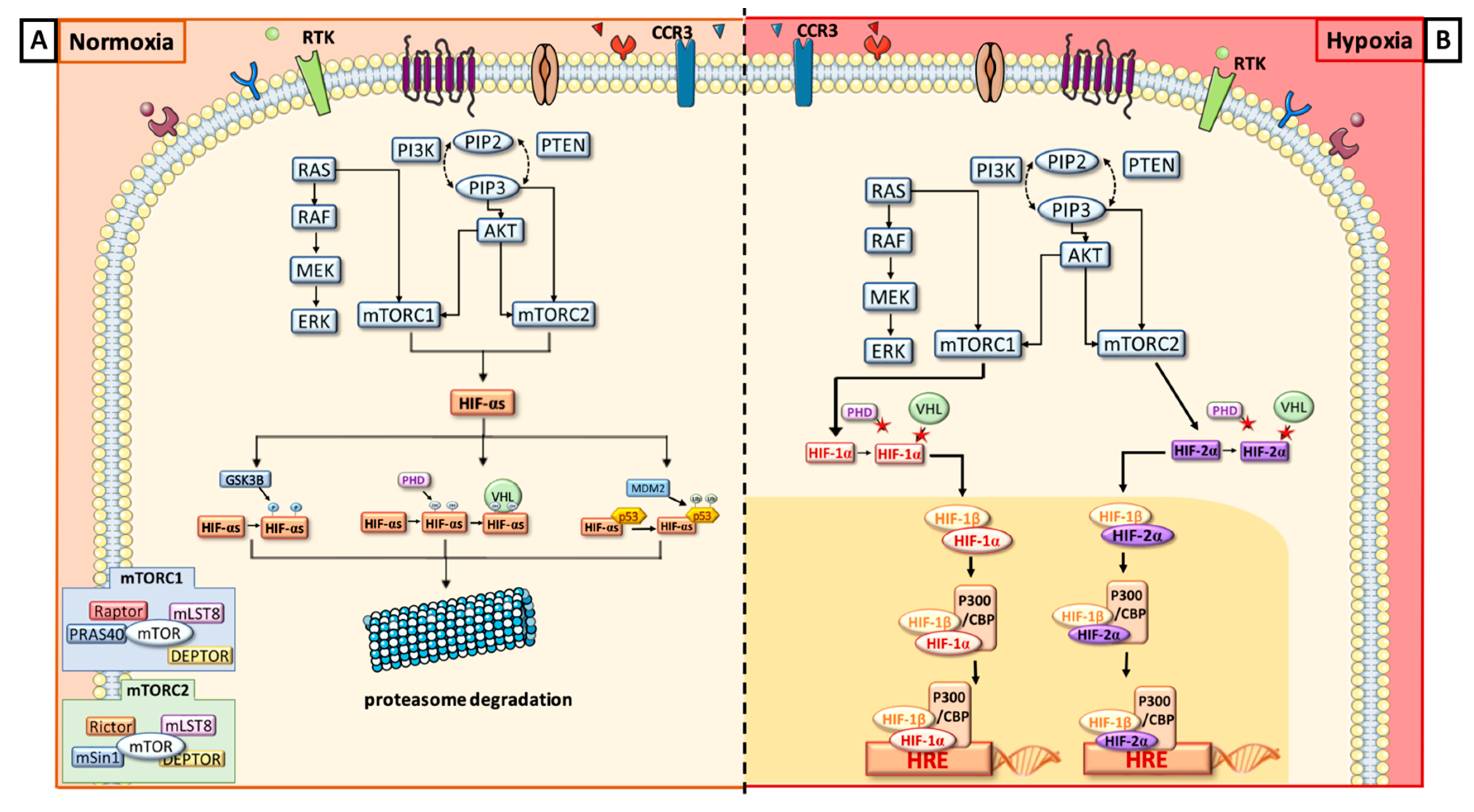{kind=link}
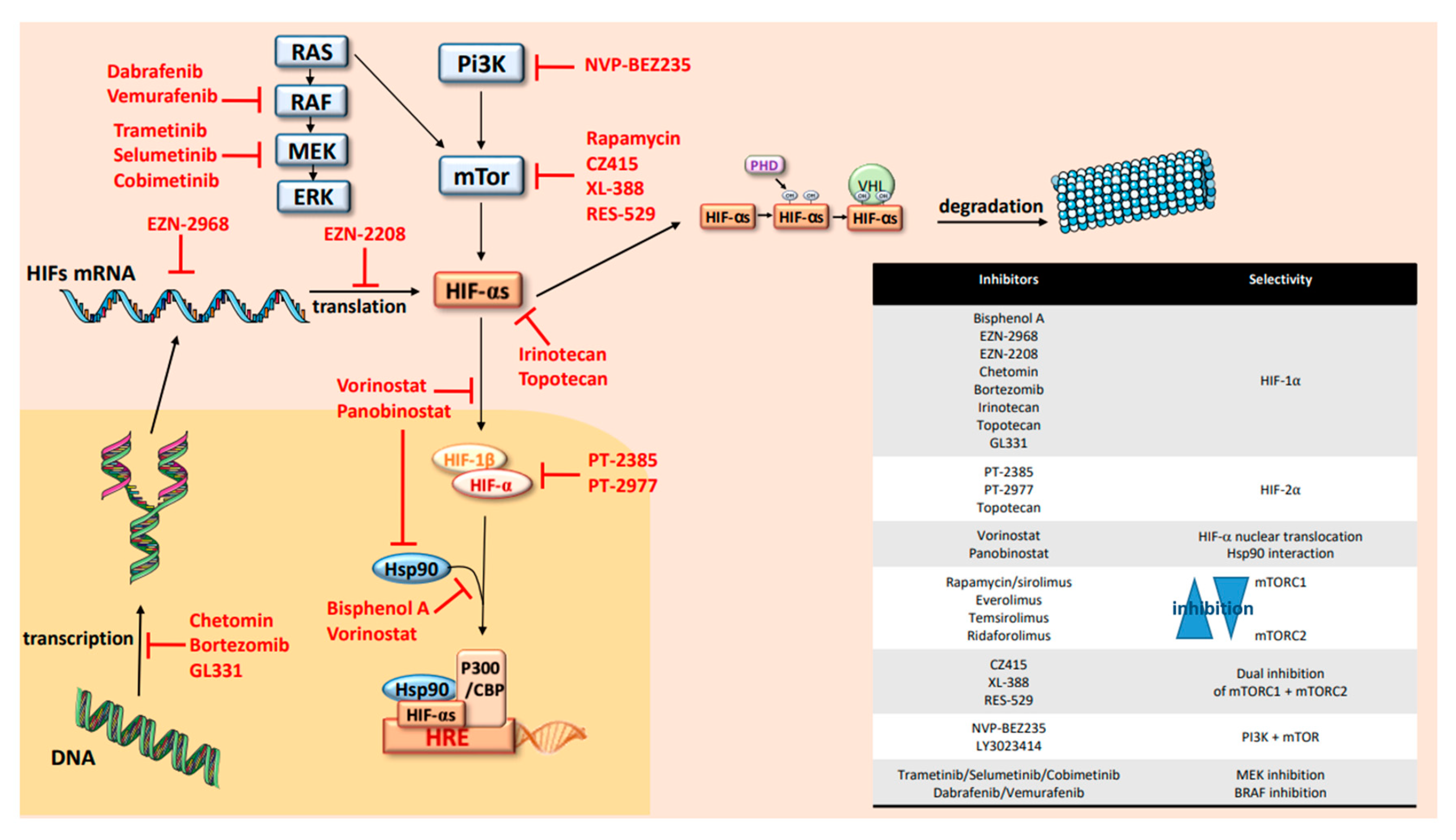{kind=link}
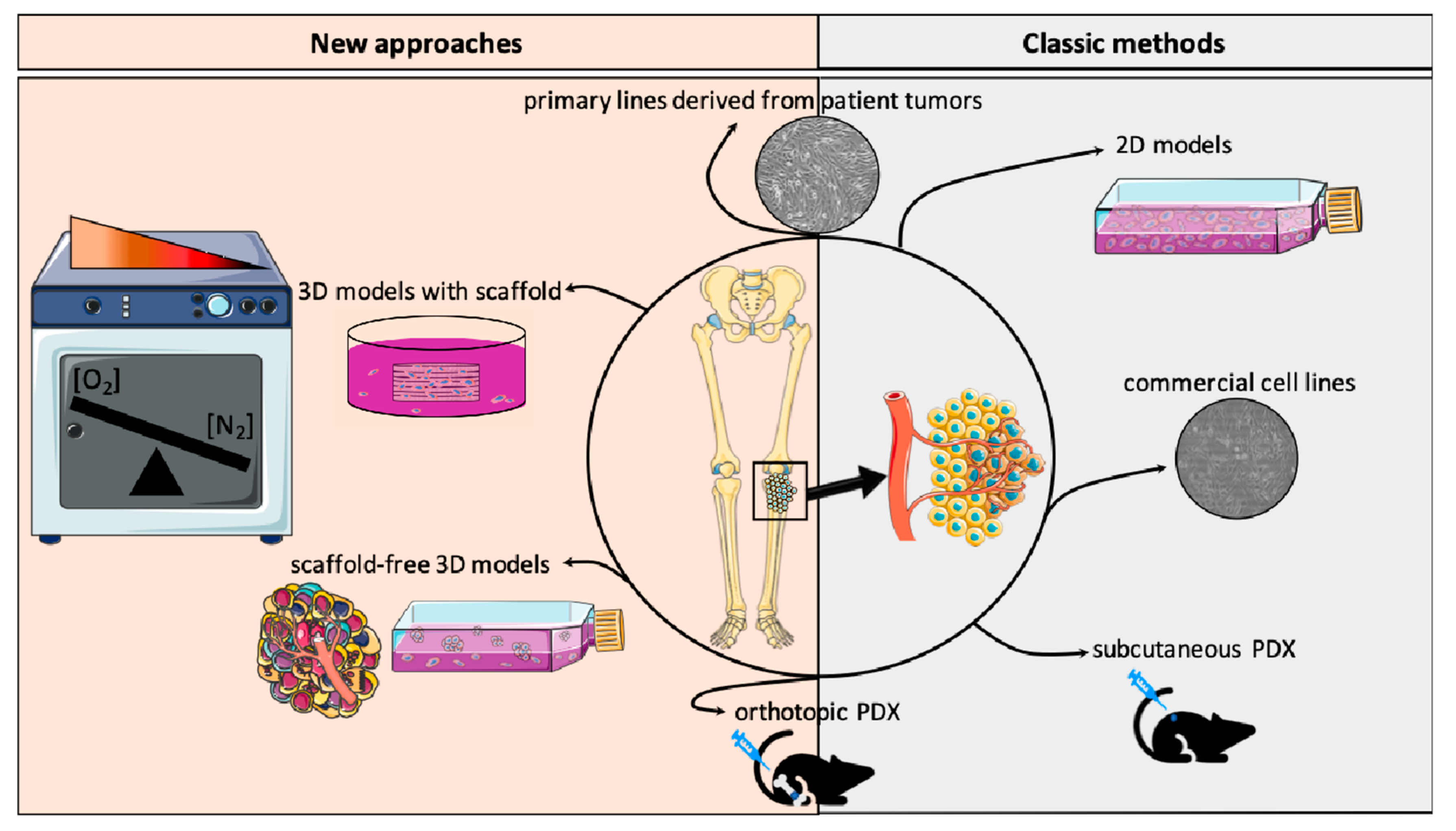{kind=link}
Abstract
:1. Introduction
2. Biomarkers Related to Hypoxia Regulation in Normal Tissues
3. Normal Bone and Hypoxia: Involvement in Osseous Production and Formation and Osteoclast-Mediated Bone Resorption
4. Presence of Hypoxic Biomarkers in Osteosarcomas Is Related to Progression and Resistance to Treatment
5. Immune Response, Hypoxia and Osteosarcoma Cells
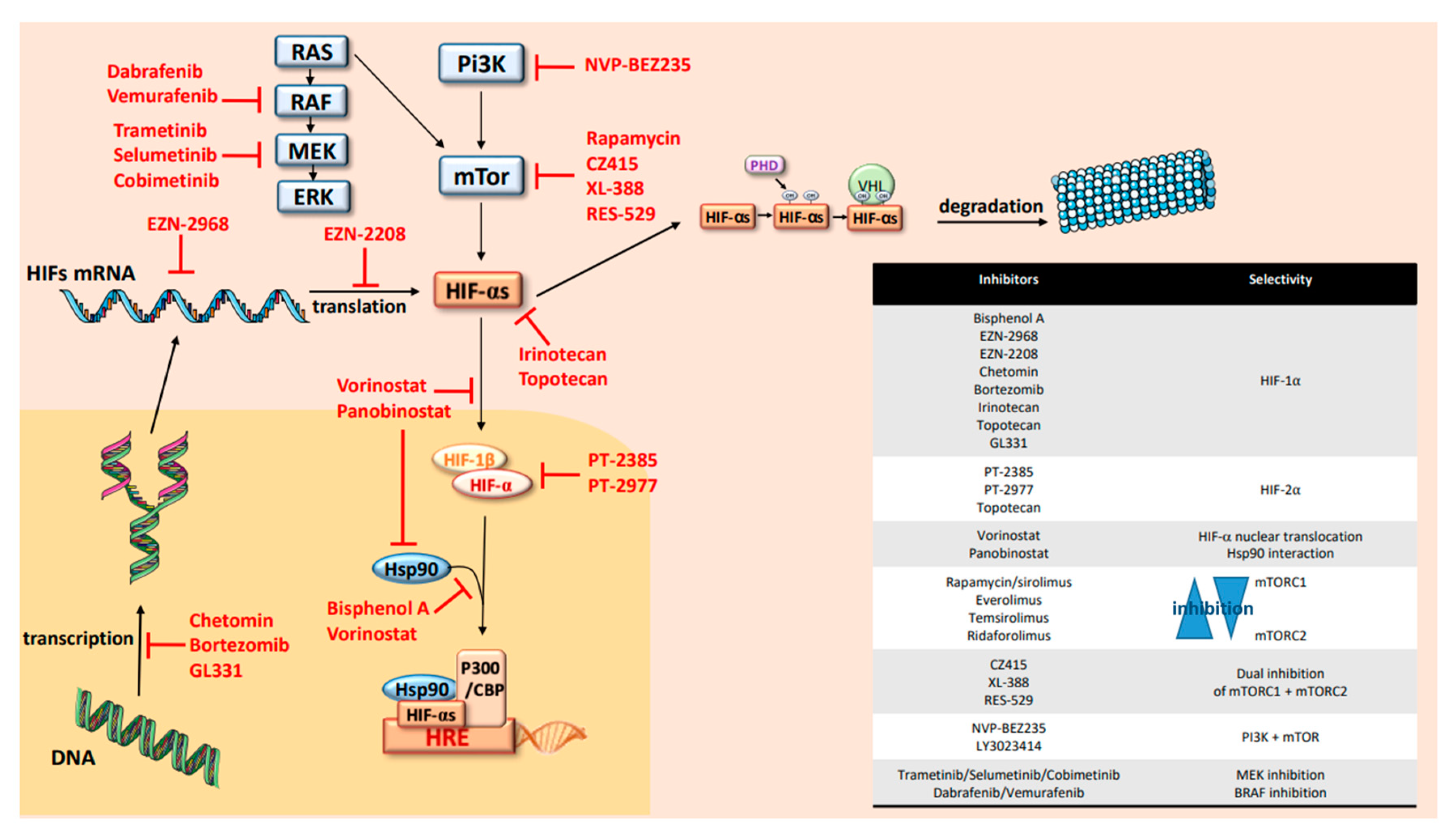
6. HIFs Targeting in OTS
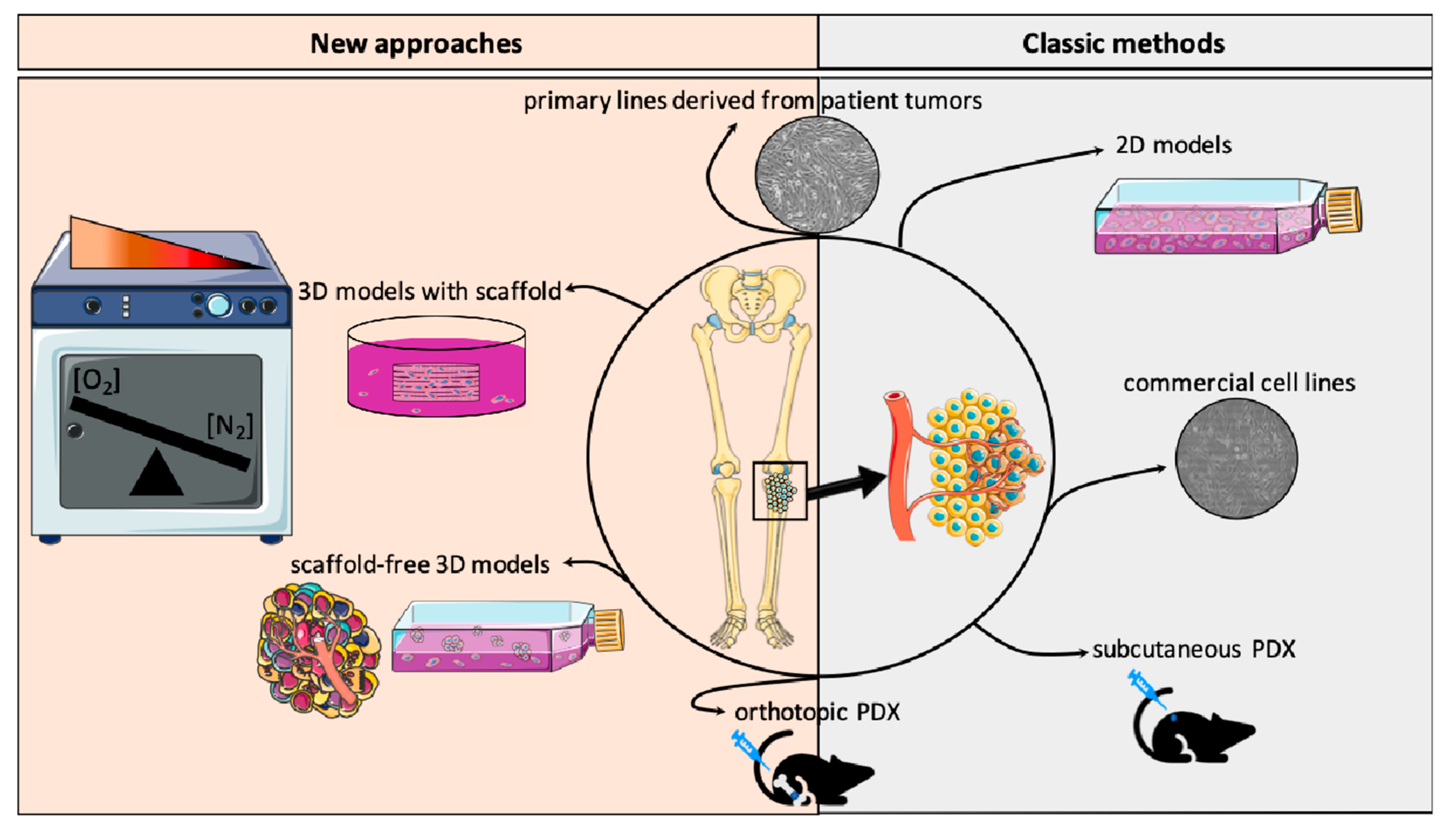
7. In Vitro and In Vivo Models to Recreate OTS Hypoxic Microenvironment
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Duong, L.M.; Richardson, L.C. Descriptive epidemiology of malignant primary osteosarcoma using population-based registries, United States, 1999–2008. J. Regist. Manag. 2013, 40, 59–64. [Google Scholar]
- Kaatsch, P.; Strothotte, J.; Becker, C.; Bielack, S.; Dirksen, U.; Blettner, M. Pediatric bone tumors in Germany from 1987 to 2011: Incidence rates, time trends and survival. Acta Oncol. 2016, 55, 1145–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchman, K.; Gao, Y.; Miller, B.T. Prognostic factors for survival in patients with high-grade osteosarcoma using the Surveillance, Epidemiology, and End Results (SEER) Program database. Cancer Epidemiol. 2015, 39, 593–599. [Google Scholar] [CrossRef]
- Moore, D.D.; Luu, H.H. Osteosarcoma. Cancer Treat. Res. 2014, 162, 65–92. [Google Scholar]
- Cates, J.M.M.; Dupont, W.D. Cytologic anaplasia is a prognostic factor in osteosarcoma biopsies, but mitotic rate or extent of spontaneous tumor necrosis are not: A critique of the College of American Pathologists Bone Biopsy template. Mod. Pathol. 2016, 30, 52–59. [Google Scholar] [CrossRef]
- Friebele, J.C.; Peck, J.; Pan, X.; Abdel-Rasoul, M.; Mayerson, J.L. Osteosarcoma: A Meta-Analysis and Review of the Literature. Am. J. Orthop. 2015, 44, 547–553. [Google Scholar]
- Zhang, T.; Kastrenopoulou, A.; Larrouture, Q.; Athanasou, N.A.; Knowles, H.J. Angiopoietin-like 4 promotes osteosarcoma cell proliferation and migration and stimulates osteoclastogenesis. BMC Cancer 2018, 18, 536. [Google Scholar] [CrossRef] [Green Version]
- Lowery, C.D.; Blosser, W.; Dowless, M.; Renschler, M.; Perez, L.V.; Stephens, J.; Pytowski, B.; Wasserstrom, H.; Stancato, L.F.; Falcon, B.; et al. Anti-VEGFR2 therapy delays growth of preclinical pediatric tumor models and enhances anti-tumor activity of chemotherapy. Oncotarget 2019, 10, 5523–5533. [Google Scholar] [CrossRef] [Green Version]
- Huvos, A.G.; Rosen, G.; Marcove, R.C. Primary osteogenic sarcoma: Pathologic aspects in 20 patients after treatment with chemotherapy en bloc resection, and prosthetic bone replacement. Arch. Pathol. Lab. Med. 1977, 101, 14–18. [Google Scholar]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahimi-Horn, M.C.; Pouysségur, J. Oxygen, a source of life and stress. FEBS Lett. 2007, 581, 3582–3591. [Google Scholar] [CrossRef]
- Wenger, R.H.; Kurtcuoglu, V.; Scholz, C.; Marti, H.H.; Hoogewijs, D. Frequently asked questions in hypoxia research. Hypoxia 2015, 3, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; LaLonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, H.; Bu, J.; Li, X.; Chen, Z.; Xiao, T. Hypoxia-Inducible Factor-1 Expression Predicts Osteosarcoma Patients’ Survival: A Meta-Analysis. Int. J. Boil. Markers 2016, 31, 229–234. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, W.; Li, P.; Tu, C. Prognosis value of Hypoxia-inducible factor-1α expression in patients with bone and soft tissue sarcoma: A meta-analysis. SpringerPlus 2016, 5, 1370. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, Y.-L.; Zhao, J.-L.; Zhen, O.; Yu, C.; Yang, B.-H.; Yu, X.-R. Hypoxia-inducible factor-1 promotes cancer progression through activating AKT/Cyclin D1 signaling pathway in osteosarcoma. Biomed. Pharmacother. 2018, 105, 1–9. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.H.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, C.W.; O’Rourke, J.F.; Nagao, M.; Gleadle, J.M.; Ratcliffe, P.J. Activation of Hypoxia-inducible Factor-1; Definition of Regulatory Domains within the α Subunit. J. Boil. Chem. 1997, 272, 11205–11214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Clin. Investig. 2007, 117, 862–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Boil. 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohlin, S.; Hamidian, A.; Von Stedingk, K.; Bridges, E.; Wigerup, C.; Bexell, D.; Påhlman, S. PI3K–mTORC2 but not PI3K–mTORC1 Regulates Transcription of HIF2A/EPAS1 and Vascularization in Neuroblastoma. Cancer Res. 2015, 75, 4617–4628. [Google Scholar] [CrossRef] [Green Version]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef] [Green Version]
- Minet, E.; Arnould, T.; Michel, G.; Roland, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett. 2000, 468, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Green, Y.S.; Sargis, T.; Reichert, E.C.; Rudasi, E.; Fuja, D.; Jonasch, E.; Koh, M.Y. Hypoxia-Associated Factor (HAF) Mediates Neurofibromin Ubiquitination and Degradation Leading to Ras–ERK Pathway Activation in Hypoxia. Mol. Cancer Res. 2019, 17, 1220–1232. [Google Scholar] [CrossRef] [Green Version]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Boil. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Using two hands to flip the angiogenic switch. Cancer Metastasis Rev. 2000, 19, 59–65. [Google Scholar] [CrossRef]
- Royds, J.A.; Dower, S.K.; Qwarnstrom, E.E.; Lewis, C.E. Response of tumour cells to hypoxia: Role of p53 and NFkB. Mol. Pathol. 1998, 51, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koka, P.; Mundre, R.S.; Rangarajan, R.; Chandramohan, Y.; Subramanian, R.; Dhanasekaran, A. Uncoupling Warburg effect and stemness in CD133+ve cancer stem cells from Saos-2 (osteosarcoma) cell line under hypoxia. Mol. Boil. Rep. 2018, 45, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Oda, T.; Nishi, K.; Takabuchi, S.; Wakamatsu, T.; Tanaka, T.; Adachi, T.; Fukuda, K.; Semenza, G.L.; Hirota, K. Macrophage Migration Inhibitory Factor Activates Hypoxia-Inducible Factor in a p53-Dependent Manner. PLoS ONE 2008, 3, e2215. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Su, X.; Liu, T. HIF-2α affects proliferation and apoptosis of MG-63 osteosarcoma cells through MAPK signaling. Mol. Med. Rep. 2017, 15, 2174–2178. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-Inducible Factor 2 Regulates Hepatic Lipid Metabolism. Mol. Cell. Boil. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [Green Version]
- Wagner, F.; Holzapfel, B.M.; Martine, L.C.; McGovern, J.; Lahr, C.A.; Boxberg, M.; Prodinger, P.M.; Grässel, S.; Loessner, D.; Hutmacher, D.W. A humanized bone microenvironment uncovers HIF2 alpha as a latent marker for osteosarcoma. Acta Biomater. 2019, 89, 372–381. [Google Scholar] [CrossRef]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Schipani, E.; Giaccia, A.J. HIF targets in bone remodeling and metastatic disease. Pharmacol. Ther. 2015, 150, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Avruch, J.; Long, X.; Lin, Y.; Ortiz-Vega, S.; Rapley, J.; Papageorgiou, A.; Oshiro, N.; Kikkawa, U. Activation of mTORC1 in two steps: Rheb-GTP activation of catalytic function and increased binding of substrates to raptor1. Biochem. Soc. Trans. 2009, 37, 223–226. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef]
- Karsenty, G. The complexities of skeletal biology. Nature 2003, 423, 316–318. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef]
- Provot, S.; Schipani, E. Molecular mechanisms of endochondral bone development. Biochem. Biophys. Res. Commun. 2005, 328, 658–665. [Google Scholar] [CrossRef]
- Cramer, T.; Schipani, E.; Johnson, R.S.; Swoboda, B.; Pfander, D. Expression of VEGF isoforms by epiphyseal chondrocytes during low-oxygen tension is HIF-1 alpha dependent. Osteoarthr. Cartil. 2004, 12, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, C.; Araldi, E.; Haigh, K.; Khatri, R.; Van Looveren, R.; Giaccia, A.J.; Haigh, J.J.; Carmeliet, G.; Schipani, E. VEGF-independent cell-autonomous functions of HIF-1α regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J. Bone Miner. Res. 2012, 27, 596–609. [Google Scholar] [CrossRef]
- Rankin, E.B.; Wu, C.; Khatri, R.; Wilson, T.L.; Andersen, R.; Araldi, E.; Rankin, A.L.; Yuan, J.; Kuo, C.J.; Schipani, E.; et al. The HIF Signaling Pathway in Osteoblasts Directly Modulates Erythropoiesis through the Production of EPO. Cell 2012, 149, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorissen, B.M.C.; De Bruin, A.; Miranda-Bedate, A.; Korthagen, N.; Wolschrijn, C.; De Vries, T.J.; Van Weeren, R.; Tryfonidou, M. Hypoxia negatively affects senescence in osteoclasts and delays osteoclastogenesis. J. Cell. Physiol. 2018, 234, 414–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulley, P.A.; Bishop, T.; Vernet, A.; Schneider, J.E.; Edwards, J.R.; Athanasou, N.A.; Knowles, H.J. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2: HIF-1α and PHD2 regulate bone resorption by osteoclasts. J. Pathol. 2017, 242, 322–333. [Google Scholar] [CrossRef]
- Li, W.; He, X.; Xue, R.; Zhang, Y.; Zhang, X.; Lu, J.; Zhang, Z.; Xue, L.; Zhan, Y. Combined over-expression of the hypoxia-inducible factor 2α gene and its long non-coding RNA predicts unfavorable prognosis of patients with osteosarcoma. Pathol.-Res. Pract. 2016, 212, 861–866. [Google Scholar] [CrossRef]
- Okuno, K.; Matsubara, T.; Nakamura, T.; Iino, T.; Kakimoto, T.; Asanuma, K.; Matsumine, A.; Sudo, A. Carbonic anhydrase IX enhances tumor cell proliferation and tumor progression in osteosarcoma. OncoTargets Ther. 2018, 11, 6879–6886. [Google Scholar] [CrossRef] [Green Version]
- Mizobuchi, H.; García-Castellano, J.M.; Philip, S.; Healey, J.H.; Gorlick, R. Hypoxia Markers in Human Osteosarcoma: An Exploratory Study. Clin. Orthop. Relat. Res. 2008, 466, 2052–2059. [Google Scholar] [CrossRef] [Green Version]
- XIX CONGRESSO NAZIONALE S.I.C.O.O.P. SOCIETA’ ITALIANA CHIRURGHI ORTOPEDICI DELL’OSPEDALITA’ PRIVATA ACCREDITATA; Capasso, L.; Florio, M.; Lillo, M.; Basilico, M.; De Santis, V.; Ziranu, A.; Grasso, A.; Minutillo, F.; Maccauro, G. Vascular endothelial growth factor expression as a biomarker of prognosis in patients with chondrosarcoma, Ewing’s sarcoma and osteosarcoma. Current concepts. J. Boil. Regul. Homeost. 2019, 33, 39–43. [Google Scholar]
- Wagner, F.; Holzapfel, B.M.; Thibaudeau, L.; Straub, M.; Ling, M.-T.; Grifka, J.; Loessner, D.; Levesque, J.-P.; Hutmacher, D.W.; Martine, L. A Validated Preclinical Animal Model for Primary Bone Tumor Research. J. Bone Joint. Surg. Am. Vol. 2016, 98, 916–925. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, S.; Chu, X.; Han, D. LncRNA HIF2PUT inhibited osteosarcoma stem cells proliferation, migration and invasion by regulating HIF2 expression. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1342–1348. [Google Scholar] [CrossRef] [Green Version]
- Bertoldo, F.; Silvestris, F.; Ibrahim, T.; Cognetti, F.; Generali, D.; Ripamonti, C.; Amadori, D.; Colleoni, M.; Conte, P.; Del Mastro, L.; et al. Targeting bone metastatic cancer: Role of the mTOR pathway. Biochim. Biophys. Acta (BBA)-Bioenergy 2014, 1845, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Yan, T.; Guo, W.; Wang, W.; Zhao, Z. Insight Into the Role of Autophagy in Osteosarcoma and Its Therapeutic Implication. Front. Oncol. 2019, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Smida, J.; Xu, H.; Zhang, Y.; Baumhoer, D.; Ribi, S.; Kovac, M.; Von Luettichau, I.; Bielack, S.; O’Leary, V.B.; Leib-Mösch, C.; et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int. J. Cancer 2017, 141, 816–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Qi, Y.-B.; Si, M.; Hou, Y.; Nie, L. A comprehensive analysis for associations between multiple microRNAs and prognosis of osteosarcoma patients. Peer J. 2020, 8, 8389. [Google Scholar]
- Fang, F.; VanCleave, A.; Helmuth, R.; Torres, H.; Rickel, K.; Wollenzien, H.; Sun, H.; Zeng, E.; Zhao, J.; Tao, J. Targeting the Wnt/β-catenin pathway in human osteosarcoma cells. Oncotarget 2018, 9, 36780–36792. [Google Scholar] [CrossRef] [Green Version]
- Scholten, D.J.; Timmer, C.M.; Peacock, J.D.; Pelle, D.W.; Williams, B.O.; Steensma, M.R. Down Regulation of Wnt Signaling Mitigates Hypoxia-Induced Chemoresistance in Human Osteosarcoma Cells. PLoS ONE 2014, 9, e111431. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Zhang, Q.; Yu, T.; Sun, S.; Wang, W.; Liu, G. Hypoxia promotes drug resistance in osteosarcoma cells via activating AMP-activated protein kinase (AMPK) signaling. J. Bone Oncol. 2016, 5, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Roncuzzi, L.; Pancotti, F.; Baldini, N. Involvement of HIF-1α activation in the doxorubicin resistance of human osteosarcoma cells. Oncol. Rep. 2014, 32, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Wu, W.; Dong, N.; Jiang, X.; Xu, J.; Zhan, X.; Zhang, Z.; Hu, Z. Mxd1 mediates hypoxia-induced cisplatin resistance in osteosarcoma cells by repression of the PTEN tumor suppressor gene. Mol. Carcinog. 2017, 56, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Kiezun, A.; Perry, J.; Tonzi, P.; Van Allen, E.; Carter, S.L.; Baca, S.; Bhatt, A.; Lawrence, M.; Walensky, L.; Wagle, N.; et al. Abstract A41: Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Sarcomas 2014, 111, 5564. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; E Kleinerman, E.; Pulé, M.; Rooney, C.M.; et al. Immunotherapy for Osteosarcoma: Genetic Modification of T cells Overcomes Low Levels of Tumor Antigen Expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.K.; Cote, G.M.; Choy, E.; Yang, P.; Harmon, D.; Schwab, J.; Nielsen, G.P.; Chebib, I.; Ferrone, S.; Wang, X.; et al. Programmed cell death 1 ligand 1 expression in osteosarcoma. Cancer Immunol. Res. 2014, 2, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J. Bone Oncol. 2019, 15, 100221. [Google Scholar] [CrossRef] [PubMed]
- Mceachron, T.A.; Triche, T.J.; Sorenson, L.; Parham, D.M.; Carpten, J.D. Profiling targetable immune checkpoints in osteosarcoma. OncoImmunology 2018, 7, e1475873-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, W.; Yuan, H.; Gu, Y.; Liu, M.; Ji, Y.; Huang, Z.; Yang, J.; Ma, L. Immune-related prognosis biomarkers associated with osteosarcoma microenvironment. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.-M.; Marie, B.; Larousserie, F.; Entz-Werlé, N.; De Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. OncoImmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; Cleton-Jansen, A.-M.; et al. Tumor-Infiltrating Macrophages Are Associated with Metastasis Suppression in High-Grade Osteosarcoma: A Rationale for Treatment with Macrophage Activating Agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Piperno-Neumann, S.; Le Deley, M.-C.; Rédini, F.; Pacquement, H.; Marec-Berard, P.; Petit, P.; Brisse, H.J.; Lervat, C.; Gentet, J.C.; Entz-Werlé, N.; et al. Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 1070–1080. [Google Scholar] [CrossRef]
- Tiedemann, K.; Le Nihouannen, D.; Fong, J.E.; Hussein, O.; Barralet, J.E.; Komarova, S.V. Regulation of Osteoclast Growth and Fusion by mTOR/raptor and mTOR/rictor/Akt. Front. Cell Dev. Boil. 2017, 5, 5. [Google Scholar] [CrossRef]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Pearce, M.C.; Jiang, Y.; Yang, L.; Goodall, C.; Miranda, C.L.; Milovancev, M.; Bracha, S.; Kolluri, S.K.; Maier, C.S. Delineation of hypoxia-induced proteome shifts in osteosarcoma cells with different metastatic propensities. Sci. Rep. 2020, 10, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2013, 73, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.E.; Shusterman, S.; Gore, L.; Muscal, J.A.; Macy, M.E.; Fox, E.; Berkowitz, N.; Buchbinder, A.; Bagatell, R. Phase 1 evaluation of EZN-2208, a polyethylene glycol conjugate of SN38, in children adolescents and young adults with relapsed or refractory solid tumors. Pediatr. Blood Cancer 2014, 61, 1792–1797. [Google Scholar] [CrossRef]
- Guérin, E.; Raffelsberger, W.; Pencreach, E.; Maier, A.; Neuville, A.; Schneider, A.; Bachellier, P.; Rohr, S.; Petitprez, A.; Poch, O.; et al. In Vivo Topoisomerase I Inhibition Attenuates the Expression of Hypoxia-Inducible Factor 1α Target Genes and Decreases Tumor Angiogenesis. Mol. Med. 2011, 18, 83–94. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Maezawa, N.; Osada, M.; Katsumura, S.; Funae, Y.; Imaoka, S. Bisphenol A, an environmental endocrine-disrupting chemical, inhibits hypoxic response via degradation of hypoxia-inducible factor 1alpha (HIF-1alpha): Structural requirement of bisphenol A for degradation of HIF-1alpha. Biochem. Biophys. Res. Commun. 2004, 318, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Aixi, Y.; Baiwen, Q.; Zonghuan, L.; Xiang, H. Inhibition of Hypoxia-inducible Factor-1 Alpha Radiosensitized MG-63 Human Osteosarcoma Cellsin Vitro. Tumori J. 2015, 101, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Abd-Aziz, N.; Stanbridge, E.J.; Shafee, N. Bortezomib attenuates HIF-1- but not HIF-2-mediated transcriptional activation. Oncol. Lett. 2015, 10, 2192–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, A.H.; Stewart, E.; Bradley, C.L.; Chen, X.; Daryani, V.; Stewart, C.F.; Calabrese, C.; Funk, A.; Miller, G.; Karlstrom, A.; et al. Combinatorial screening using orthotopic patient derived xenograft-expanded early phase cultures of osteosarcoma identify novel therapeutic drug combinations. Cancer Lett. 2019, 442, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-R.; Zhou, X.-Z.; Zhu, L.-Q.; Yao, C.; Fang, J.-F.; Zhou, F.; Deng, X.-W.; Zhang, Y.-Q. The anti-cancer activity of the mTORC1/2 dual inhibitor XL388 in preclinical osteosarcoma models. Oncotarget 2016, 7, 49527–49538. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-C.; Cui, Z.-F.; Chen, S.-M.; Yang, L.-J.; Lian, H.-K.; Liu, B.; Su, Z.-H.; Liu, J.-S.; Wang, M.; Hu, Z.-B.; et al. NVP-BEZ235 synergizes cisplatin sensitivity in osteosarcoma. Oncotarget 2017, 9, 10483–10496. [Google Scholar] [CrossRef]
- Perut, F.; Carta, F.; Bonuccelli, G.; Grisendi, G.; Di Pompo, G.; Avnet, S.; Sbrana, F.V.; Hosogi, S.; Dominici, M.; Kusuzaki, K.; et al. Carbonic anhydrase IX inhibition is an effective strategy for osteosarcoma treatment. Expert Opin. Ther. Targets 2015, 19, 1593–1605. [Google Scholar] [CrossRef]
- Cortini, M.; Baldini, N.; Avnet, S. New Advances in the Study of Bone Tumors: A Lesson From the 3D Environment. Front. Physiol. 2019, 10, 814. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, M.V.; Gaspar, V.M.; Ferreira, L.P.; Mano, J.F. Hydrogel 3D in vitro tumor models for screening cell aggregation mediated drug response. Biomater. Sci. 2020, 8, 1855–1864. [Google Scholar] [CrossRef]
- Castillo-Tandazo, W.; Mutsaers, A.J.; Walkley, C.R. Osteosarcoma in the Post Genome Era: Preclinical Models and Approaches to Identify Tractable Therapeutic Targets. Curr. Osteoporos. Rep. 2019, 17, 343–352. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierrevelcin, M.; Fuchs, Q.; Lhermitte, B.; Messé, M.; Guérin, E.; Weingertner, N.; Martin, S.; Lelong-Rebel, I.; Nazon, C.; Dontenwill, M.; et al. Focus on Hypoxia-Related Pathways in Pediatric Osteosarcomas and Their Druggability. Cells 2020, 9, 1998. https://doi.org/10.3390/cells9091998
Pierrevelcin M, Fuchs Q, Lhermitte B, Messé M, Guérin E, Weingertner N, Martin S, Lelong-Rebel I, Nazon C, Dontenwill M, et al. Focus on Hypoxia-Related Pathways in Pediatric Osteosarcomas and Their Druggability. Cells. 2020; 9(9):1998. https://doi.org/10.3390/cells9091998
Chicago/Turabian StylePierrevelcin, Marina, Quentin Fuchs, Benoit Lhermitte, Melissa Messé, Eric Guérin, Noelle Weingertner, Sophie Martin, Isabelle Lelong-Rebel, Charlotte Nazon, Monique Dontenwill, and et al. 2020. "Focus on Hypoxia-Related Pathways in Pediatric Osteosarcomas and Their Druggability" Cells 9, no. 9: 1998. https://doi.org/10.3390/cells9091998
APA StylePierrevelcin, M., Fuchs, Q., Lhermitte, B., Messé, M., Guérin, E., Weingertner, N., Martin, S., Lelong-Rebel, I., Nazon, C., Dontenwill, M., & Entz-Werlé, N. (2020). Focus on Hypoxia-Related Pathways in Pediatric Osteosarcomas and Their Druggability. Cells, 9(9), 1998. https://doi.org/10.3390/cells9091998