Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer


Abstract
1. Introduction
2. Development of MDSCs
2.1. VEGF-Driven MDSC Development
2.2. G-CSF and GM-CSF-Driven MDSC Development
2.3. Cytokine-Driven MDSC Development
3. Accumulation of MDSCs to Tumor Sites
4. Immunosuppressive Functions of MDSCs
4.1. ROS, iNOS, and ARG-1
4.2. Anti-Inflammatory Cytokine Production, Exosomes, and Immune Checkpoint Regulation
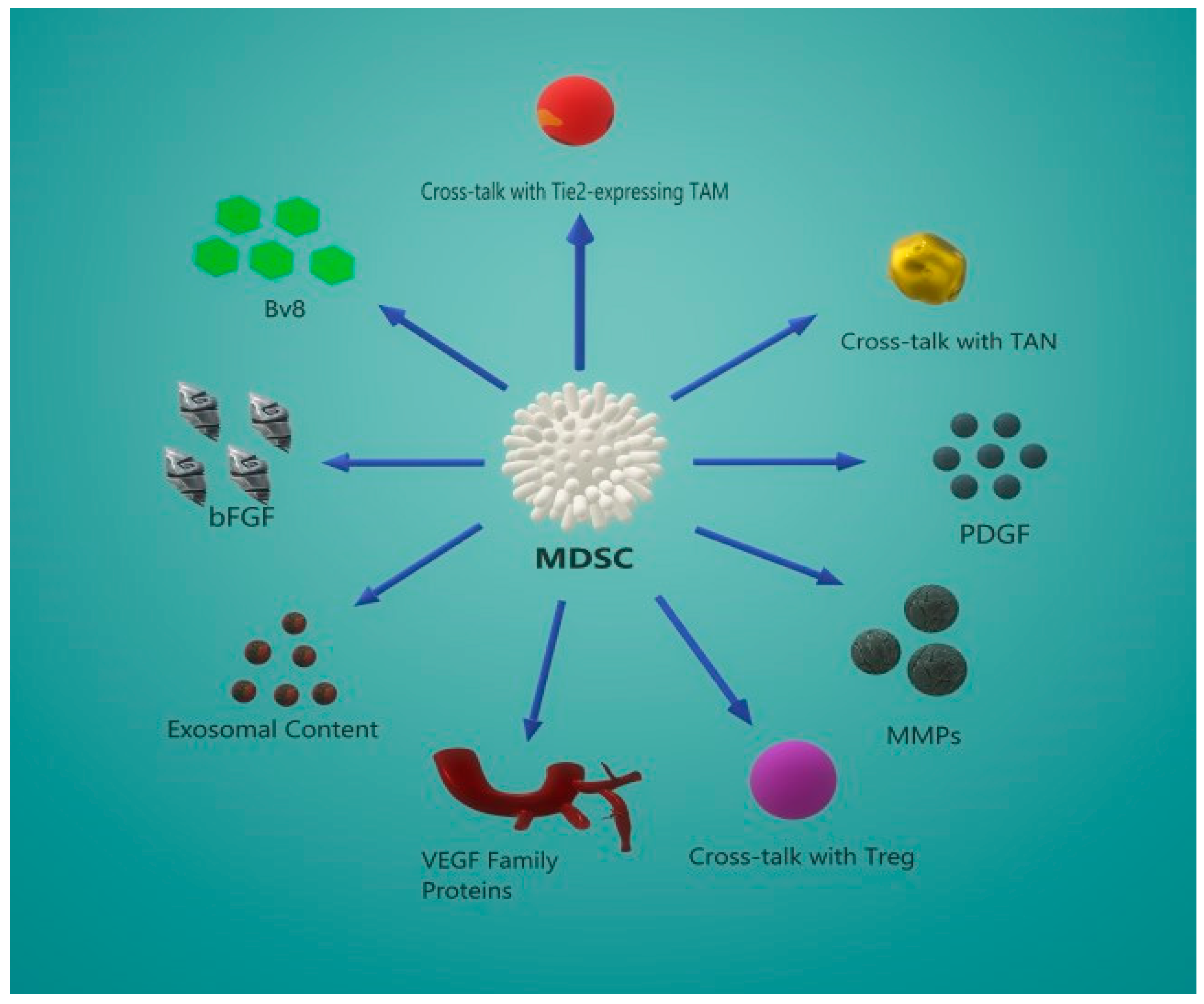
5. MDSC-Induced Angiogenesis
5.1. The VEGF/VEGFR Angiogenic Pathway
5.2. Secondary Angiogenic Mechanisms
5.3. Crosstalk Between MDSCs and Other Effector Cells
5.4. MDSC-Derived Exosome Content Promotes Angiogenesis
6. MDSC Targeting for Cancer Therapy
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human tumor-infiltrating myeloid cells: Phenotypic and functional diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced: MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Wong, J.L.; Edwards, R.P.; Odunsi, K.; Moysich, K.; Kalinski, P. PGE 2-Driven Induction and Maintenance of Cancer-Associated Myeloid-Derived Suppressor Cells. Immunol. Investig. 2012, 41, 635–657. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Poschke, I.; Kiessling, R. On the armament and appearances of human myeloid-derived suppressor cells. Clin. Immunol. 2012, 144, 250–268. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef]
- Kim, C. Homeostatic and pathogenic extramedullary hematopoiesis. J. Blood Med. 2010, 1, 13–19. [Google Scholar] [CrossRef]
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649–3665. [Google Scholar] [CrossRef]
- Mao, Y.; Poschke, I.; Wennerberg, E.; De Coaña, Y.P.; Brage, S.E.; Schultz, I.; Hansson, J.; Masucci, G.; Lundqvist, A.; Kiessling, R. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 2013, 73, 3877–3887. [Google Scholar] [CrossRef]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef] [PubMed]
- Damuzzo, V.; Pinton, L.; Desantis, G.; Solito, S.; Marigo, I.; Bronte, V.; Mandruzzato, S. Complexity and Challenges in Defining Myeloid-Derived Suppressor Cells. Cytom. Part B Clin. Cytom. 2015, 88, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Koinis, F.; Gioulbasani, M.; Aggouraki, D.; Koutoulaki, A.; Skalidaki, E.; Mavroudis, D.; Georgoulias, V.; Kotsakis, A. A circulating subpopulation of monocytic myeloid-derived suppressor cells as an independent prognostic/predictive factor in untreated non-small lung cancer patients. J. Immunol. Res. 2014, 2014, 659294. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Beatty, P.L.; McKolanis, J.; Brand, R.; Schoen, R.E.; Finn, O.J. Circulating Myeloid Derived Suppressor Cells (MDSC) That Accumulate in Premalignancy Share Phenotypic and Functional Characteristics With MDSC in Cancer. Front. Immunol. 2019, 10, 1401. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age review-article. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key criminals of tumor pre-metastatic niche formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef]
- Pilatova, K.; Bencsikova, B.; Demlova, R.; Valik, D.; Zdrazilova-Dubska, L. Myeloid-derived suppressor cells (MDSCs) in patients with solid tumors: Considerations for granulocyte colony-stimulating factor treatment. Cancer Immunol. Immunother. 2018, 67, 1919–1929. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Safi, S.; Blattner, C.; Rathinasamy, A.; Umansky, L.; Juenger, S.; Warth, A.; Eichhorn, M.; Muley, T.; Herth, F.J.F.; et al. Circulating and tumor myeloid-derived suppressor cells in resectable non-small cell lung cancer. Am. J. Respir. Crit. Care Med. 2018, 198, 777–787. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv. Cancer Res. 2015, 128, 95–139. [Google Scholar] [PubMed]
- Zöller, M. Janus-faced myeloid-derived suppressor cell exosomes for the good and the bad in cancer and autoimmune disease. Front. Immunol. 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Koinis, F.; Vetsika, E.K.; Aggouraki, D.; Skalidaki, E.; Koutoulaki, A.; Gkioulmpasani, M.; Georgoulias, V.; Kotsakis, A. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, 1263–1272. [Google Scholar] [CrossRef]
- Abrams, S.I.; Waight, J.D. Identification of a G-CSF-Granulocytic MDSC axis that promotes tumor progression. Oncoimmunology 2012, 1, 550–551. [Google Scholar] [CrossRef][Green Version]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Gomez, A.F.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, T.; Shao, S.; Shi, B.; Zhao, Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1004983. [Google Scholar] [CrossRef]
- Lee, C.R.; Lee, W.; Cho, S.K.; Park, S.G. Characterization of multiple cytokine combinations and TGF-β on differentiation and functions of myeloid-derived suppressor cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef] [PubMed]
- Ding, A.S.; Routkevitch, D.; Jackson, C.; Lim, M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front. Immunol. 2019, 10, 1715. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Liu, Z.; Zhang, J.; Jin, X. Myeloid-derived suppressor cell accumulation in renal cell carcinoma is correlated with CCL2, IL-17 and IL-18 expression in blood and tumors. Adv. Clin. Exp. Med. 2018, 27, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, C.; Sanmamed, M.F.; Rodríguez-Ruiz, M.E.; Teijeira, Á.; Oñate, C.; González, Á.; Ponz, M.; Schalper, K.A.; Pérez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef]
- Chouaib, S.; Umansky, V.; Kieda, C. The role of hypoxia in shaping the recruitment of proangiogenic and immunosuppressive cells in the tumor microenvironment. Contemp. Oncol. 2018, 22, 7–13. [Google Scholar] [CrossRef]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J. Hematol. Oncol. 2019, 12, 84–102. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, Z.; Wu, L.; Zhang, M.; Li, W.; Ding, J.; Zhu, J.; Wei, H.; Zhao, K. Circulating and Tumor-Infiltrating Myeloid-Derived Suppressor Cells in Patients with Colorectal Carcinoma. PLoS ONE 2013, 8, e57114. [Google Scholar] [CrossRef]
- Shou, D.; Wen, L.; Song, Z.; Yin, J.; Sun, Q.; Gong, W. Suppressive role of myeloid-derived suppressor cells (MDSCs) in the microenvironment of breast cancer and targeted immunotherapies. Oncotarget 2016, 7, 64505–64511. [Google Scholar] [CrossRef]
- Ibáñez-Vea, M.; Zuazo, M.; Gato, M.; Arasanz, H.; Fernández-Hinojal, G.; Escors, D.; Kochan, G. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment: Current Knowledge and Future Perspectives. Arch. Immunol. Ther. Exp. (Warsz) 2018, 66, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T cells sustain myeloid-derived suppressor cell mediated immune suppression. Oncotarget 2016, 7, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Kondo, Y.; Kimura, O.; Morosawa, T.; Fujisaka, Y.; Umetsu, T.; Kogure, T.; Inoue, J.; Nakagome, Y.; Shimosegawa, T. PD-L1+ MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci. Rep. 2016, 6, 39296. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef]
- De Haas, N.; de Koning, C.; Spilgies, L.; de Vries, I.J.M.; Hato, S.V. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology 2016, 5, e1196312. [Google Scholar] [CrossRef]
- Cimen Bozkus, C.; Elzey, B.D.; Crist, S.A.; Ellies, L.G.; Ratliff, T.L. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J. Immunol. 2015, 195, 5237–5250. [Google Scholar] [CrossRef]
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014, 74, 5723–5733. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Kisin, E.R.; Yanamala, N.; Tkach, A.V.; Gutkin, D.W.; Star, A.; Shurin, G.V.; Kagan, V.E.; Shurin, M.R. MDSC and TGFβ are required for facilitation of tumor growth in the lungs of mice exposed to carbon nanotubes. Cancer Res. 2015, 75, 1615–1623. [Google Scholar] [CrossRef]
- Geis-Asteggiante, L.; Belew, A.T.; Clements, V.K.; Edwards, N.J.; Ostrand-Rosenberg, S.; El-Sayed, N.M.; Fenselau, C. Differential Content of Proteins, mRNAs, and miRNAs Suggests that MDSC and Their Exosomes May Mediate Distinct Immune Suppressive Functions. J. Proteome Res. 2018, 17, 486–498. [Google Scholar] [CrossRef]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 Promotes Tumor Development through the Induction of Tumor Promoting Microenvironments at Tumor Sites and Myeloid-Derived Suppressor Cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef]
- Zhu, X.; Mulcahy, L.A.; Mohammed, R.A.; Lee, A.H.; Franks, H.A.; Kilpatrick, L.; Yilmazer, A.; Paish, E.C.; Ellis, I.O.; Patel, P.M.; et al. IL-17 expression by breast-cancer-associated macrophages: IL-17 promotes invasiveness of breast cancer cell lines. Breast Cancer Res. 2008, 10, R95. [Google Scholar] [CrossRef] [PubMed]
- Mucha, J.; Majchrzak, K.; Taciak, B.; Hellmén, E.; Król, M. MDSCs mediate angiogenesis and predispose canine mammary tumor cells for metastasis via IL-28/IL-28RA (IFN-λ) signaling. PLoS ONE 2014, 9, e103249. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1: PD-L1/2 pathway from discovery to clinical implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef] [PubMed]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Moughon, D.; Orpilla, J.R.; Shin, N.P.; Sedighim, S.; Treger, J.; Odesa, S.; Tucker, A.; et al. Immunosuppressive tumor-infltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro-Oncology 2017, 19, 796–807. [Google Scholar]
- Rivera, L.B.; Bergers, G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol. 2015, 36, 240–249. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef]
- Johnson, B.W.; Achyut, B.R.; Fulzele, S.; Mondal, A.K.; Kolhe, R.; Arbab, A.S. Delineating pro-angiogenic myeloid cells in cancer therapy. Int. J. Mol. Sci. 2018, 19, 2565. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-L.; Banerjee, S.; White, S.; Kortylewski, M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int. J. Mol. Sci. 2018, 19, 1803. [Google Scholar] [CrossRef]
- Liu, J.-F.; Deng, W.-W.; Chen, L.; Li, Y.-C.; Wu, L.; Ma, S.-R.; Zhang, W.-F.; Bu, L.-L.; Sun, Z.-J. Inhibition of JAK2/STAT3 reduces tumor-induced angiogenesis and myeloid-derived suppressor cells in head and neck cancer. Mol. Carcinog. 2018, 57, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Li, J.; Qu, P.; Lin, P.C. C/EBP-δ positively regulates MDSC expansion and endothelial VEGFR2 expression in tumor development. Oncotarget 2017, 8, 50582–50593. [Google Scholar] [CrossRef] [PubMed]
- Binsfeld, M.; Muller, J.; Lamour, V.; De Veirman, K.; De Raeve, H.; Bellahcène, A.; Van Valckenborgh, E.; Baron, F.; Beguin, Y.; Caers, J.; et al. Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget 2016, 7, 37931–37943. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Yen, M.C.; Chang, W.A.; Tsai, P.H.; Pan, Y.C.; Liao, S.H.; Kuo, P.L. CXCL17-derived CD11b+Gr-1+ myeloid-derived suppressor cells contribute to lung metastasis of breast cancer through platelet-derived growth factor-BB. Breast Cancer Res. 2019, 21, 23–36. [Google Scholar] [CrossRef]
- Fahey, E.; Doyle, S.L. IL-1 family cytokine regulation of vascular permeability and angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef] [PubMed]
- Pingwara, R.; Witt-Jurkowska, K.; Ulewicz, K.; Mucha, J.; Tonecka, K.; Pilch, Z.; Taciak, B.; Zabielska-Koczywas, K.; Mori, M.; Berardozzi, S.; et al. Interferon lambda 2 promotes mammary tumor metastasis via angiogenesis extension and stimulation of cancer cell migration. J. Physiol. Pharmacol. 2017, 68, 573–583. [Google Scholar] [PubMed]
- Deryugina, E.I.; Zajac, E.; Juncker-Jensen, A.; Kupriyanova, T.A.; Welter, L.; Quigley, J.P. Tissue-Infiltrating Neutrophils Constitute the Major In Vivo Source of Angiogenesis-Inducing MMP-9 in the Tumor Microenvironment. Neoplasia 2014, 16, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef]
- Szebeni, G.J.; Vizler, C.; Nagy, L.I.; Kitajka, K.; Puskas, L.G. Pro-tumoral inflammatory myeloid cells as emerging therapeutic targets. Int. J. Mol. Sci. 2016, 17, 1958. [Google Scholar] [CrossRef]
- Guedez, L.; Jensen-Taubman, S.; Bourboulia, D.; Kwityn, C.J.; Wei, B.; Caterina, J.; Stetler-Stevenson, W.G. TIMP-2 targets tumor-associated myeloid suppressor cells with effects in cancer immune dysfunction and angiogenesis. J. Immunother. 2012, 35, 502–512. [Google Scholar] [CrossRef]
- Hossain, F.; Majumder, S.; Ucar, D.A.; Rodriguez, P.C.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Notch signaling in myeloid cells as a regulator of tumor immune responses. Front. Immunol. 2018, 9, 1288. [Google Scholar] [CrossRef]
- Sammarco, G.; Varricchi, G.; Ferraro, V.; Ammendola, M.; De Fazio, M.; Altomare, D.F.; Luposella, M.; Maltese, L.; Currò, G.; Marone, G.; et al. Mast cells, angiogenesis and lymphangiogenesis in human gastric cancer. Int. J. Mol. Sci. 2019, 20, 2106. [Google Scholar] [CrossRef]
- Burke, M.; Choksawangkarn, W.; Edwards, N.; Ostrand-Rosenberg, S.; Fenselau, C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J. Proteome Res. 2014, 13, 836–843. [Google Scholar] [CrossRef]
- Deng, Z.; Rong, Y.; Teng, Y.; Zhuang, X.; Samykutty, A.; Mu, J.; Zhang, L.; Cao, P.; Yan, J.; Miller, D.; et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene 2017, 36, 639–651. [Google Scholar] [CrossRef]
- Liu, Q.; Peng, F.; Chen, J. The Role of Exosomal MicroRNAs in the Tumor Microenvironment of Breast Cancer. Int. J. Mol. Sci. 2019, 20, 3884. [Google Scholar] [CrossRef] [PubMed]
- Leija Montoya, G.; González Ramírez, J.; Sandoval Basilio, J.; Serafín Higuera, I.; Isiordia Espinoza, M.; González González, R.; Serafín Higuera, N. Long Non-coding RNAs: Regulators of the Activity of Myeloid-Derived Suppressor Cells. Front. Immunol. 2019, 10, 1734. [Google Scholar] [CrossRef] [PubMed]
- Botti, G.; Scognamiglio, G.; Aquino, G.; Liguori, G.; Cantile, M. LncRNA HOTAIR in tumor microenvironment: What role? Int. J. Mol. Sci. 2019, 20, 2279. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Combinatorial Approaches with Checkpoint Inhibitors to Enhance Anti-tumor Immunity. Front. Immunol. 2019, 10, 999. [Google Scholar] [CrossRef]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef]
- Lu, L.-C.; Chang, C.-J.; Hsu, C.-H. Targeting myeloid-derived suppressor cells in the treatment of hepatocellular carcinoma: Current state and future perspectives. J. Hepatocell. Carcinoma 2019, 6, 71–84. [Google Scholar] [CrossRef]
- Bosiljcic, M.; Cederberg, R.A.; Hamilton, M.J.; LePard, N.E.; Harbourne, B.T.; Collier, J.L.; Halvorsen, E.C.; Shi, R.; Franks, S.E.; Kim, A.Y.; et al. Targeting myeloid-derived suppressor cells in combination with primary mammary tumor resection reduces metastatic growth in the lungs. Breast Cancer Res. 2019, 21, 103. [Google Scholar] [CrossRef]
- Bauer, R.; Udonta, F.; Wroblewski, M.; Ben-Batalla, I.; Santos, I.M.; Taverna, F.; Kuhlencord, M.; Gensch, V.; Päsler, S.; Vinckier, S.; et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 2018, 78, 3220–3232. [Google Scholar]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27–38. [Google Scholar] [CrossRef]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18. [Google Scholar] [CrossRef]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-Associated macrophages in glioma neo-Angiogenesis and implications for anti-Angiogenic strategies. Neuro-Oncology 2017, 19, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Penn, C.A.; Yang, K.; Zong, H.; Lim, J.Y.; Cole, A.; Yang, D.; Baker, J.; Goonewardena, S.N.; Buckanovich, R.J. Therapeutic impact of nanoparticle therapy targeting tumor-associated macrophages. Mol. Cancer Ther. 2018, 17, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, L.M.; Fritsch, M.; Gebauer, F.; Günther, S.D.; Stair, N.R.; Seeger, J.M.; Thangarajah, F.; Dieplinger, G.; Bludau, M.; Alakus, H.; et al. Tumour-infiltrating neutrophils counteract anti-VEGF therapy in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Park, S.Y.; Henry, V.; Smith, B.D.; Tiao, N.; Flynn, D.L.; De Groot, J.F. Novel MET/TIE2/VEGFR2 inhibitor altiratinib inhibits tumor growth and invasiveness in bevacizumab-resistant glioblastoma mouse models. Neuro-Oncology 2016, 18, 1230–1241. [Google Scholar] [CrossRef]


{kind=link}
| Mechanism | Mediated By | Effect |
|---|---|---|
| Induction of immunosuppressive cells | Release of IFN-γ, IL-10, and TGF-β | Induction of Tregs |
| Release of IL-10 | Generation of M2 macrophages | |
| Impaired lymphocyte homing | Cleavage of L-selectin by the metalloprotease ADAM 17 | Reduction in the homing and antigen-dependent activation of CD8+ T cells in lymph nodes |
| Downregulation of CD44 and P-selectin by NO-producing M-MDSC | Blocking of T cell extravasation and tissue infiltration | |
| Production of reactive oxygen species (ROS) | NADPH oxidase 2 (NOX-2) | Reduced CD3ζ-chain expression Inhibition of T cell proliferation Increase of ARG1 expression |
| Nitric oxide production | Induction of COX-2 expression Induction of HIF-1α expression Increase of ARG1 expression | Induction of T cell anergy |
| Induction of nitrogen species | Induction of T cell apoptosis TCR nitration Chemokine nitration | |
| Cysteine/cystine and L-arginine deprivation | Increased uptake of L-arginine by the CAT2B transporter | Reduced TCR ζ-chain expression Inhibition of T cell proliferation Increase of ARG1 |
| Increased uptake of cysteine via SLC7A11 transporter | Reduced protein synthesis Glutathione production | |
| Adenosine production | Induction of the ectoenzymes CD39 and CD73 via TGFβ and hypoxia | Decreased phosphorylation of Zap70, ERK, and Akt Reduced expression of CD95L, perforin, IFN-γ, TNF-α, CD25 in T cells |
| Activation of immuno-regulatory molecules | High expression of B7 | T cell anergy |
| High expression of PD-L1 | T cell apoptosis | |
| High expression of FasL | Upregulation of Fas receptor |
| Title | Malignancy | Treatment | Trial No | Phase |
|---|---|---|---|---|
| A Study of RGX-104 in Patients with Advanced Solid Malignancies and Lymphoma | Malignant neoplasms | RGX-104; Nivolumab Ipilimumab; Docetaxel Pembrolizumab Carboplatin Pemetrexed | NCT02922764 | Phase I |
| Trial to Evaluate Safety and Efficacy of Vinorelbine with Metronomic Administration in Combination with Atezolizumab as Second-line Treatment for Patients with Stage IV Non-small Cell Lung Cancer (VinMetAtezo) | Non-small cell lung cancer | Atezolizumab Vinorelbine | NCT03801304 | Phase II |
| Dendritic Cell Vaccine with or Without Gemcitabine Pre-Treatment for Adults and Children with Sarcoma | Sarcoma | Dendritic Cells Vaccine | NCT01803152 | Phase I |
| Soft tissue sarcoma | Lysate of Tumor | |||
| Bone sarcoma | Gemcitabine Imiquimod | |||
| Capecitabine + Bevacizumab in Patients with Recurrent Glioblastoma | Glioblastoma | Capecitabine Bevacizumab | NCT02669173 | Phase I |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vetsika, E.-K.; Koukos, A.; Kotsakis, A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells 2019, 8, 1647. https://doi.org/10.3390/cells8121647
Vetsika E-K, Koukos A, Kotsakis A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells. 2019; 8(12):1647. https://doi.org/10.3390/cells8121647
Chicago/Turabian StyleVetsika, Eleni-Kyriaki, Aristeidis Koukos, and Athanasios Kotsakis. 2019. "Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer" Cells 8, no. 12: 1647. https://doi.org/10.3390/cells8121647
APA StyleVetsika, E.-K., Koukos, A., & Kotsakis, A. (2019). Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells, 8(12), 1647. https://doi.org/10.3390/cells8121647