Critical Impact of Human Amniotic Membrane Tension on Mitochondrial Function and Cell Viability In Vitro

 ,
, 
 , , and
, , and 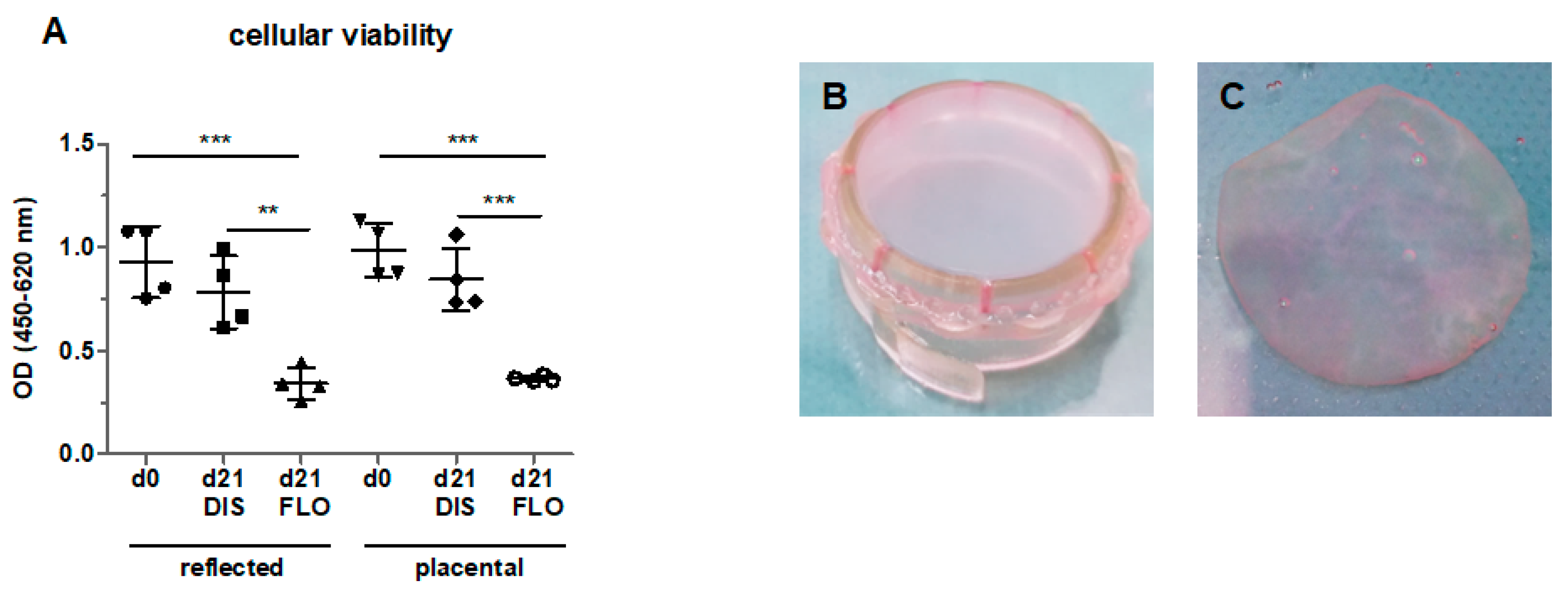{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of the Human Amniotic Membrane (hAM)
2.2. Cultivation of hAM Samples
2.3. Cell Viability Assay
2.4. Laser Scanning Confocal Microscopy
2.5. High Resolution Respirometry
2.6. ATP Measurement
2.7. Histology
2.8. Transmission Electron Microscopy
2.9. Reverse-Transcription Quantitative PCR Analysis
2.10. Statistical Analysis
3. Results and Discussion
3.1. Cellular Viability Strongly Decreased in Floating hAM Samples
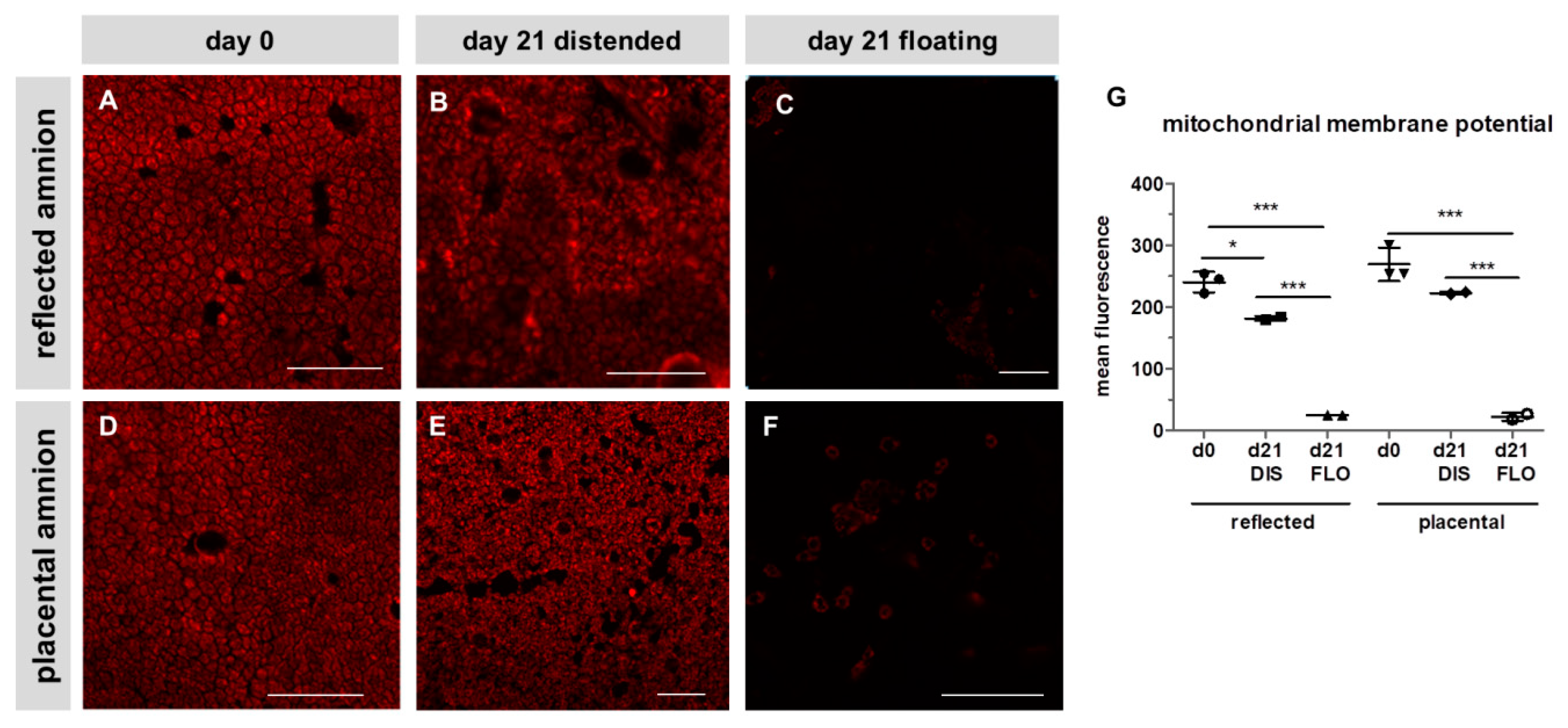
3.2. Mitochondrial Membrane Potential Drastically Decreased in Floating hAM Samples
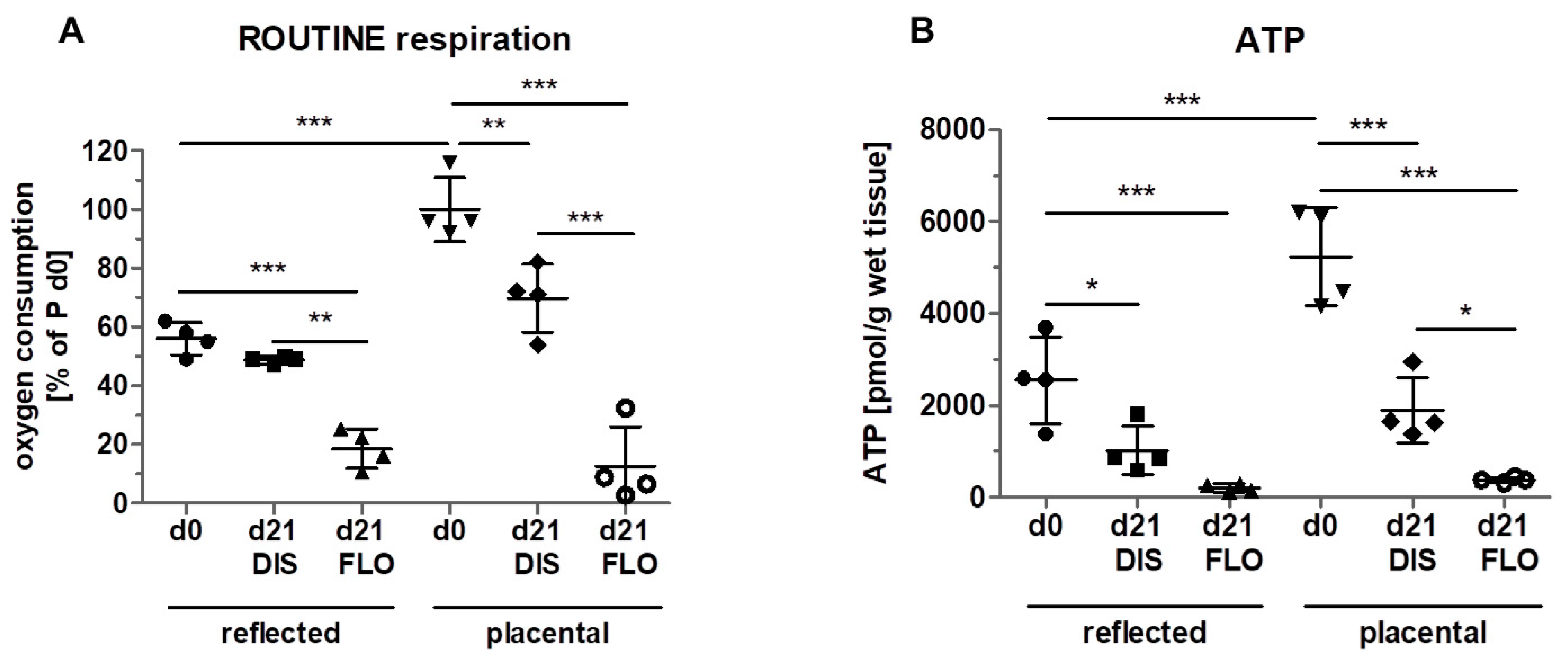
3.3. Mitochondrial Respiration and ATP Concentration Strongly Decreased in Floating hAM Samples
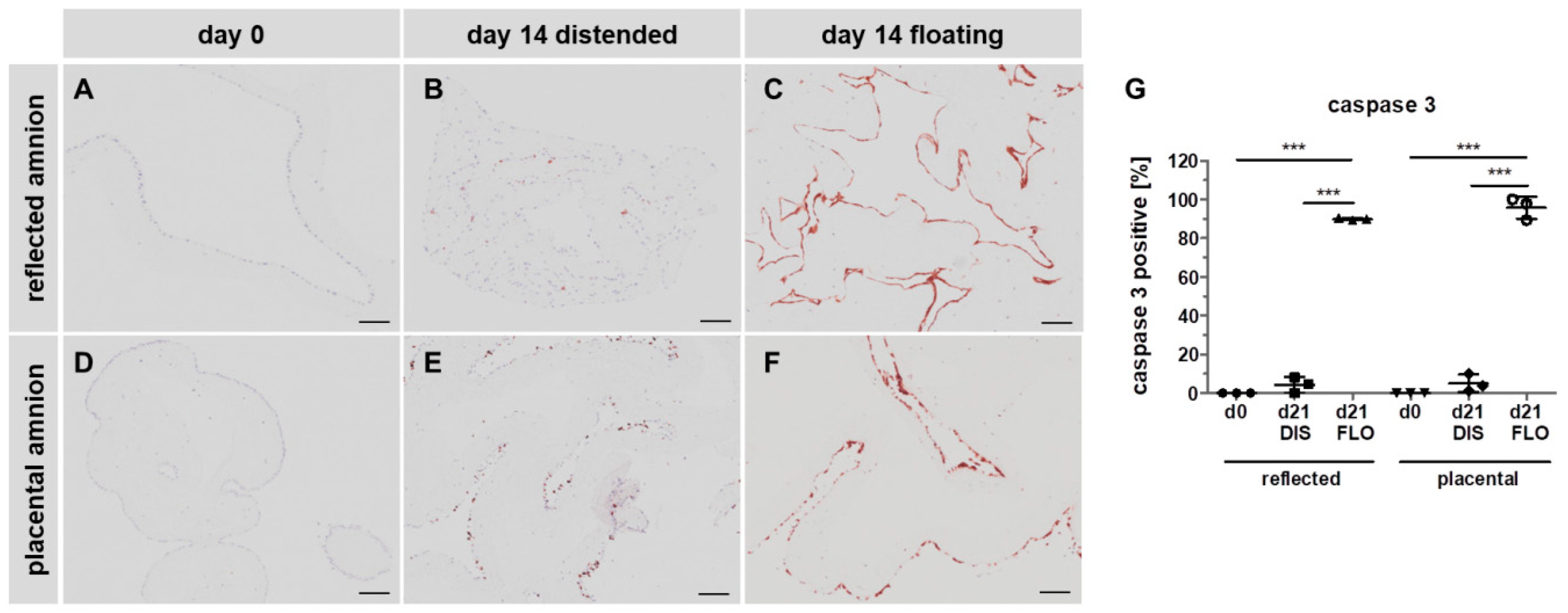
3.4. Caspase 3 is Strongly Upregulated in Floating hAM Samples
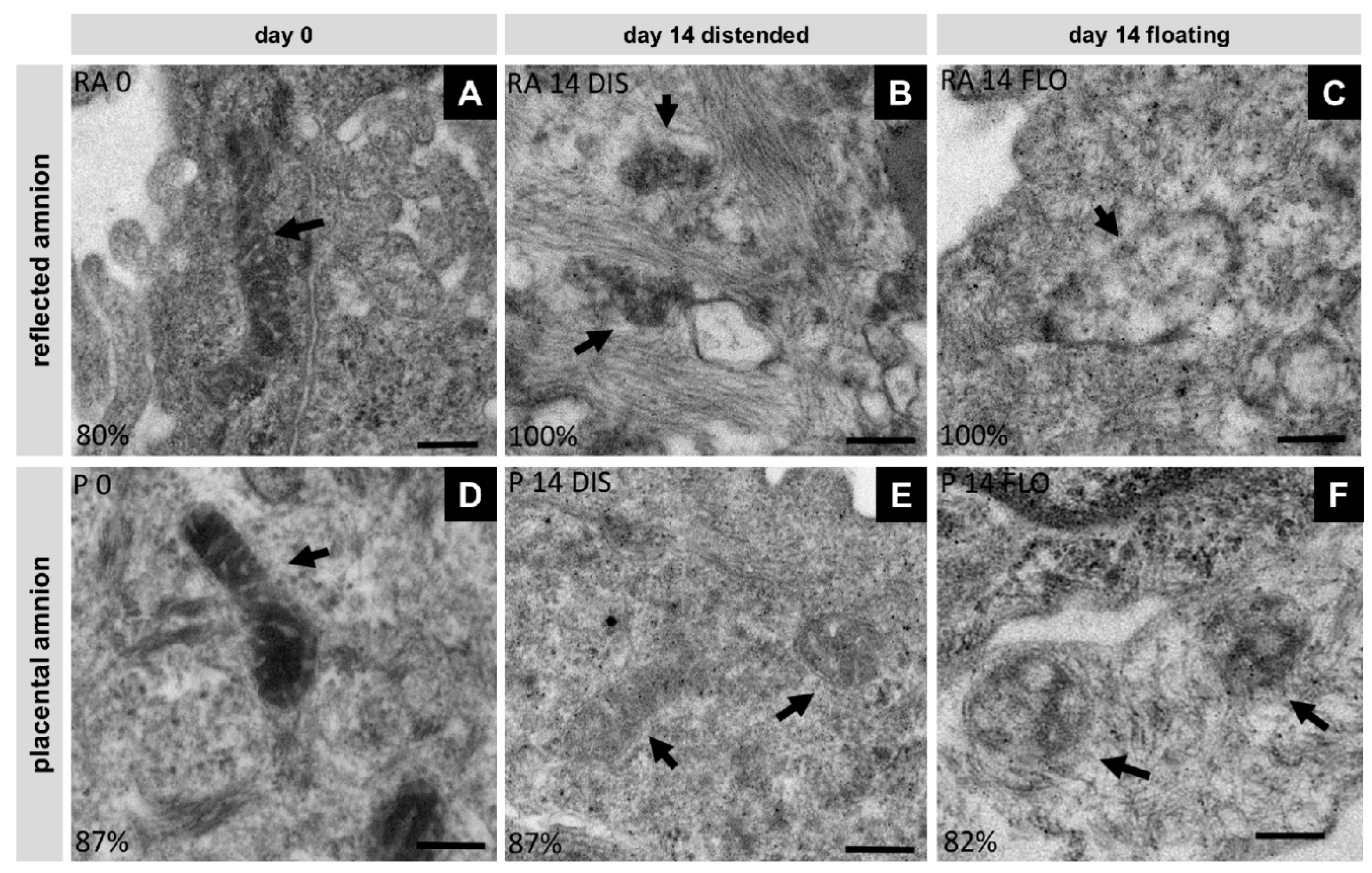
3.5. Severe Loss of Mitochondrial Internal Structure under Floating Conditions
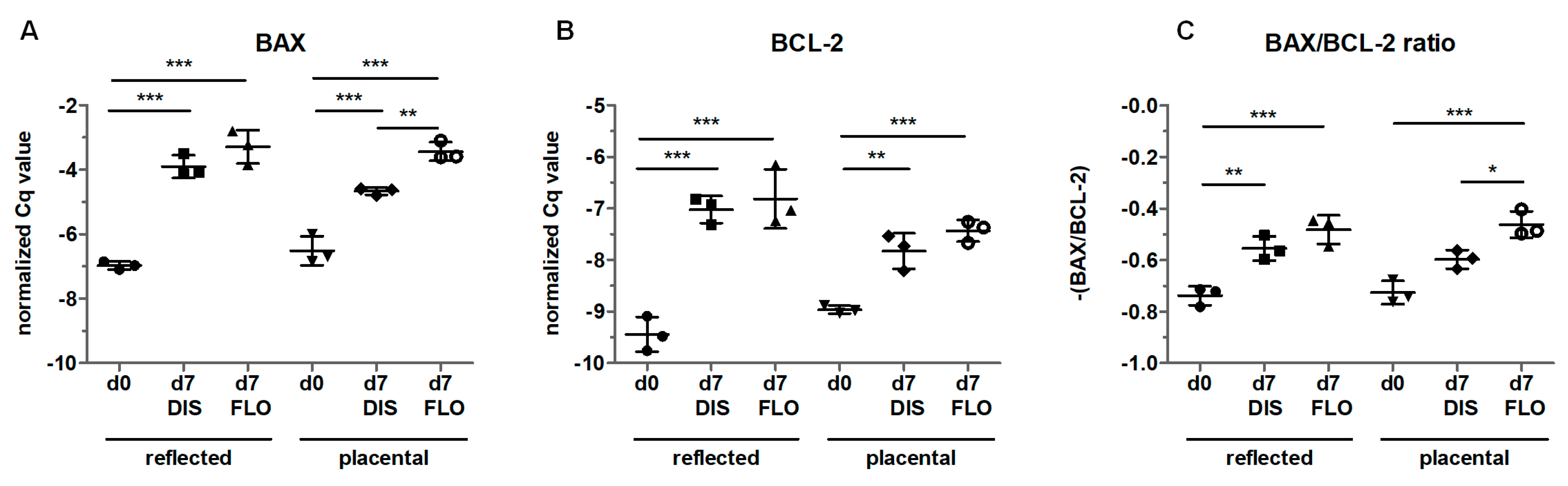
3.6. Mitochondria-Linked Apoptotic Gene Expression was Upregulated within Seven Days
4. Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stern, M. The Grafting of Preserved Amniotic Membrane to Burned and Ulcerated Surfaces, substituting Skin Grafts. J. Am. Med. Assoc. 1912, 60, 973–974. [Google Scholar] [CrossRef]
- Sabella, N. Use of Fetal Membranes in Skin Grafting. Med. Rec. 1913, 83, 478–480. [Google Scholar]
- Rötth, A. De PLASTIC REPAIR OF CONJUNCTIVAL DEFECTS WITH FETAL MEMBRANES. Arch. Ophthalmol. 1940, 23, 522–525. [Google Scholar] [CrossRef]
- Rohleder, N.H.; Loeffelbein, D.J.; Feistl, W.; Eddicks, M.; Wolff, K.D.; Gulati, A.; Steinstraesser, L.; Kesting, M.R. Repair of oronasal fistulae by interposition of multilayered amniotic membrane allograft. Plast. Reconstr. Surg. 2013, 132, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Silini, A.R.; Cargnoni, A.; Magatti, M.; Pianta, S.; Parolini, O. The Long Path of Human Placenta, and Its Derivatives, in Regenerative Medicine. Front. Bioeng. Biotechnol. 2015, 3, 162. [Google Scholar] [CrossRef] [PubMed]
- Sadler, T.W.; Langman, J. Langman’s Medical Embryology, 9th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Sakuragawa, N.; Enosawa, S.; Ishii, T.; Thangavel, R.; Tashiro, T.; Okuyama, T.; Suzuki, S. Human amniotic epithelial cells are promising transgene carriers for allogeneic cell transplantation into liver. J. Hum. Genet. 2000, 45, 171–176. [Google Scholar] [CrossRef]
- Portmann-Lanz, C.B.; Schoeberlein, A.; Huber, A.; Sager, R.; Malek, A.; Holzgreve, W.; Surbek, D.V. Placental mesenchymal stem cells as potential autologous graft for pre- and perinatal neuroregeneration. Am. J. Obstet. Gynecol. 2006, 194, 664–673. [Google Scholar] [CrossRef]
- Lindenmair, A.; Wolbank, S.; Stadler, G.; Meinl, A.; Peterbauer-Scherb, A.; Eibl, J.; Polin, H.; Gabriel, C.; van Griensven, M.; Redl, H. Human Amniotic Membrane: Osteogenic Differentiation in toto. Biomaterials 2010, 31, 8659–8665. [Google Scholar] [CrossRef]
- Banerjee, A.; Nürnberger, S.; Hennerbichler, S.; Riedl, S.; Schuh, C.M.A.P.; Hacobian, A.; Teuschl, A.; Eibl, J.; Redl, H.; Wolbank, S. In toto differentiation of human amniotic membrane towards the Schwann cell lineage. Cell Tissue Bank. 2014, 15, 227–239. [Google Scholar] [CrossRef]
- Miki, T.; Mitamura, K.; Ross, M.A.; Stolz, D.B.; Strom, S.C. Identification of stem cell marker-positive cells by immunofluorescence in term human amnion. J. Reprod. Immunol. 2007, 75, 91–96. [Google Scholar] [CrossRef]
- Bailo, M.; Soncini, M.; Vertua, E.; Signoroni, P.B.; Sanzone, S.; Lombardi, G.; Arienti, D.; Calamani, F.; Zatti, D.; Paul, P.; et al. Engraftment Potential of Human Amnion and Chorion Cells Derived from Term Placenta. Transplantation 2004, 78, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Niederkorn, J.Y.; Neelam, S.; Mayhew, E.; Word, R.A.; McCulley, J.P.; Alizadeh, H. Immunosuppressive factors secreted by human amniotic epithelial cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Wolbank, S.; Peterbauer, A.; Fahrner, M.; Hennerbichler, S.; van Griensven, M.; Stadler, G.; Redl, H.; Gabriel, C. Dose-dependent immunomodulatory effect of human stem cells from amniotic membrane: a comparison with human mesenchymal stem cells from adipose tissue. Tissue Eng. 2007, 13, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Kronsteiner, B.; Wolbank, S.; Peterbauer, A.; Hackl, C.; Redl, H.; van Griensven, M.; Gabriel, C. Human Mesenchymal Stem Cells from Adipose Tissue and Amnion Influence T-Cells Depending on Stimulation Method and Presence of Other Immune Cells. Stem Cells Dev. 2011, 20, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.M.; Romero, R.; Kim, J.-S.; Tarca, A.L.; Kim, S.K.; Draghici, S.; Kusanovic, J.P.; Gotsch, F.; Mittal, P.; Hassan, S.S.; et al. Region-Specific Gene Expression Profiling: Novel Evidence for Biological Heterogeneity of the Human Amnion1. Biol. Reprod. 2008, 79, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Romero, R.; Tarca, A.L.; Bhatti, G.; Lee, J.H.; Chaiworapongsa, T.; Hassan, S.S.; Kim, C.J. MiR-143 regulation of prostaglandin-endoperoxidase synthase 2 in the amnion: Implications for human parturition at term. PLoS ONE 2011, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Weidinger, A.; Hofer, M.; Steinborn, R.; Lindenmair, A.; Hennerbichler-Lugscheider, S.; Eibl, J.; Redl, H.; Kozlov, A.V.; Wolbank, S. Different metabolic activity in placental and reflected regions of the human amniotic membrane. Placenta 2015, 36, 1329–1332. [Google Scholar] [CrossRef]
- Litwiniuk, M.; Radowicka, M.; Krejner, A.; Śladowska, A.; Grzela, T. Amount and distribution of selected biologically active factors in amniotic membrane depends on the part of amnion and mode of childbirth. Can we predict properties of amnion dressing? A proof-of-concept study. Cent.-Eur. J. Immunol 2017, 42, 1–6. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S. Mitochondrial transfer from bone marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2013, 18, 759–765. [Google Scholar] [CrossRef]
- Liu, C.-S.; Chang, J.-C.; Kuo, S.-J.; Liu, K.-H.; Lin, T.-T.; Cheng, W.-L.; Chuang, S.-F. Delivering healthy mitochondria for the therapy of mitochondrial diseases and beyond. Int. J. Biochem. Cell Biol. 2014, 53, 141–146. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Kruse, F.E.; Joussen, A.M.; Rohrschneider, K.; You, L.; Sinn, B.; Baumann, J.; Volcker, H.E. Cryopreserved human amniotic membrane for ocular surface reconstruction. Graefes Arch. Clin. Exp. Ophthalmol. 2000, 238, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Lindenmair, A.; Nürnberger, S.; Stadler, G.; Meinl, A.; Hackl, C.; Eibl, J.; Gabriel, C.; Hennerbichler, S.; Redl, H.; Wolbank, S. Intact human amniotic membrane differentiated towards the chondrogenic lineage. Cell Tissue Bank. 2014, 15, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Skinner, S.J.; Campos, G.A.; Liggins, G.C. Collagen content of human amniotic membranes: Effect of gestation length and premature rupture. Obstet. Gynecol. 1981, 57, 487–489. [Google Scholar] [PubMed]
- Parry, S.; Strauss, J.F. Premature Rupture of the Fetal Membranes. N. Engl. J. Med. 1998, 338, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Millar, L.K.; Stollberg, J.; DeBuque, L.; Bryant-Greenwood, G. Fetal membrane distention: Determination of the intrauterine surface area and distention of the fetal membranes preterm and at term. Am. J. Obstet. Gynecol. 2000, 182, 128–134. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Banerjee, A.; Lindenmair, A.; Hennerbichler, S.; Steindorf, P.; Steinborn, R.; Kozlov, A.V.; Redl, H.; Wolbank, S.; Weidinger, A. Cellular and Site-Specific Mitochondrial Characterization of Vital Human Amniotic Membrane. Cell Transplant. 2018, 27, 3–11. [Google Scholar] [CrossRef]
- Banerjee, A.; Lindenmair, A.; Steinborn, R.; Dumitrescu, S.D.; Hennerbichler, S.; Kozlov, A.V.; Redl, H.; Wolbank, S.; Weidinger, A. Oxygen Tension Strongly Influences Metabolic Parameters and the Release of Interleukin-6 of Human Amniotic Mesenchymal Stromal Cells In vitro. Stem Cells Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Malak, T.M.; Bell, S.C. Structural characteristics of term human fetal membranes: A novel zone of extreme morphological alteration within the rupture site. Br. J. Obstet. Gynaecol. 1994, 101, 375–386. [Google Scholar] [CrossRef]
- Saikumar, P.; Dong, Z.; Patel, Y.; Hall, K.; Hopfer, U.; Weinberg, J.M.; Venkatachalam, M.A. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene 1998, 17, 3401–3415. [Google Scholar] [CrossRef] [PubMed]
- Latta, M.; Künstle, G.; Leist, M.; Wendel, A. Metabolic depletion of ATP by fructose inversely controls CD95- and tumor necrosis factor receptor 1-mediated hepatic apoptosis. J. Exp. Med. 2000, 191, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef]
- Seo, A.Y.; Joseph, A.-M.M.; Dutta, D.; Hwang, J.C.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, S.J.; Menon, R.; Bryant, C.; Lombardi, S.J. Programmed cell death (apoptosis) as a possible pathway to metalloproteinase activation and fetal membrane degradation in premature rupture of membranes. Am. J. Obstet. Gynecol. 2000, 182, 1468–1476. [Google Scholar] [CrossRef]
- Shen, Z.Y.; Li, E.M.; Lu, S.Q.; Shen, J.; Cai, Y.M.; Wu, Y.E.; Zheng, R.M.; Tan, L.J.; Xu, L.Y. Autophagic and Apoptotic Cell Death in Amniotic Epithelial Cells. Placenta 2008, 29, 956–961. [Google Scholar] [CrossRef]
- Kendal-Wright, C.E.; Hubbard, D.; Bryant-Greenwood, G.D. Chronic Stretching of Amniotic Epithelial Cells Increases Pre-B Cell Colony-Enhancing Factor (PBEF/Visfatin) Expression and Protects Them from Apoptosis. Placenta 2008, 29, 255–265. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poženel, L.; Lindenmair, A.; Schmidt, K.; Kozlov, A.V.; Grillari, J.; Wolbank, S.; Banerjee, A.; Weidinger, A. Critical Impact of Human Amniotic Membrane Tension on Mitochondrial Function and Cell Viability In Vitro. Cells 2019, 8, 1641. https://doi.org/10.3390/cells8121641
Poženel L, Lindenmair A, Schmidt K, Kozlov AV, Grillari J, Wolbank S, Banerjee A, Weidinger A. Critical Impact of Human Amniotic Membrane Tension on Mitochondrial Function and Cell Viability In Vitro. Cells. 2019; 8(12):1641. https://doi.org/10.3390/cells8121641
Chicago/Turabian StylePoženel, Laura, Andrea Lindenmair, Katy Schmidt, Andrey V. Kozlov, Johannes Grillari, Susanne Wolbank, Asmita Banerjee, and Adelheid Weidinger. 2019. "Critical Impact of Human Amniotic Membrane Tension on Mitochondrial Function and Cell Viability In Vitro" Cells 8, no. 12: 1641. https://doi.org/10.3390/cells8121641
APA StylePoženel, L., Lindenmair, A., Schmidt, K., Kozlov, A. V., Grillari, J., Wolbank, S., Banerjee, A., & Weidinger, A. (2019). Critical Impact of Human Amniotic Membrane Tension on Mitochondrial Function and Cell Viability In Vitro. Cells, 8(12), 1641. https://doi.org/10.3390/cells8121641