The Effect of Fibroblast Growth Factor 15 Signaling in Non-Steatotic and Steatotic Liver Transplantation from Cardiocirculatory Death

 , , , ,
, , , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Cardiocirculatory Death Induction
2.3. Surgical Procedure of Liver Transplantation
2.4. Experimental Design
2.5. Biochemical Assays
2.6. Western Blotting
2.7. Reverse Transcription and Quantitative Polymerase Chain Reaction
2.8. Liver and Intestine Histology
2.9. Immunohistochemistry
2.10. Statistics
3. Results
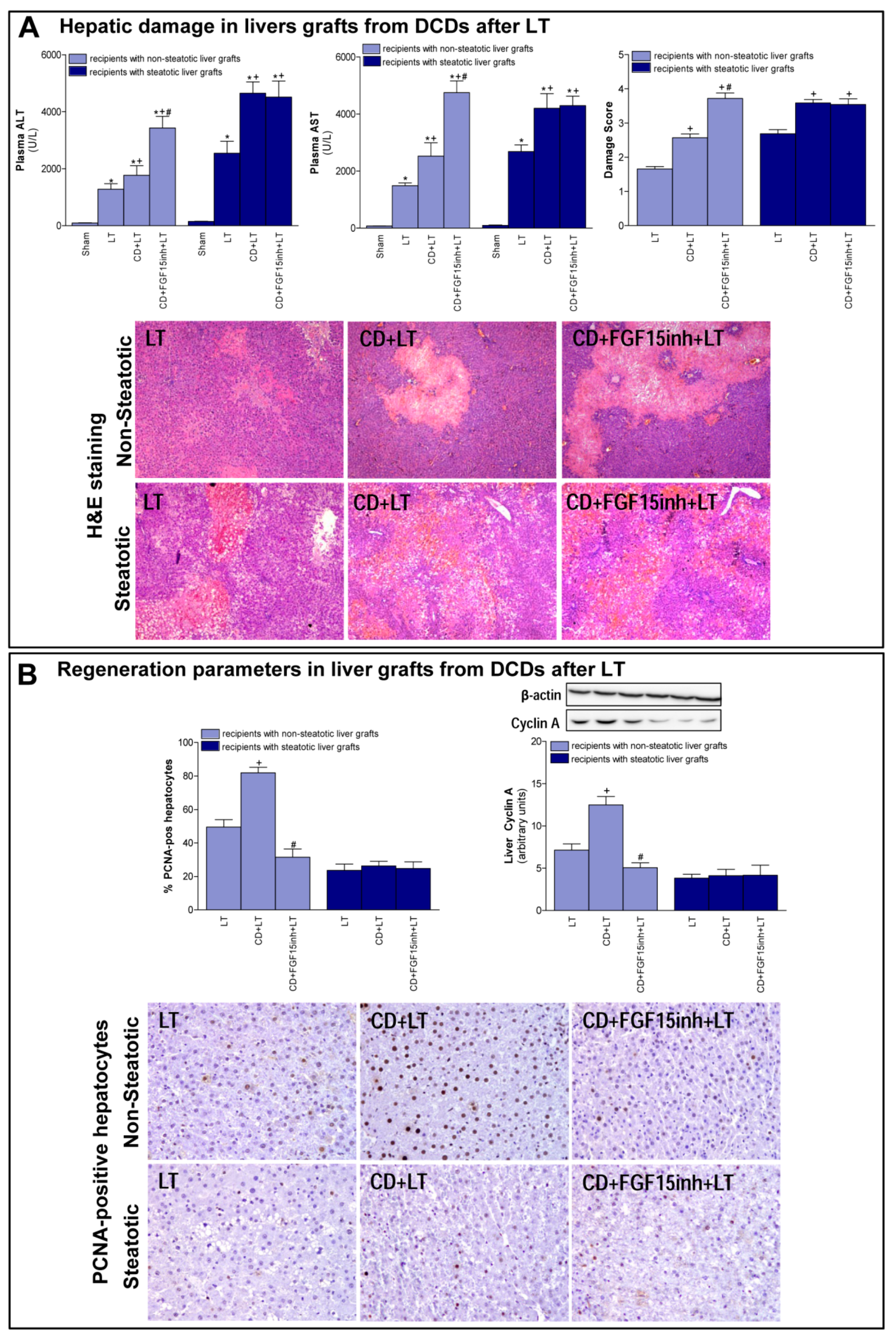
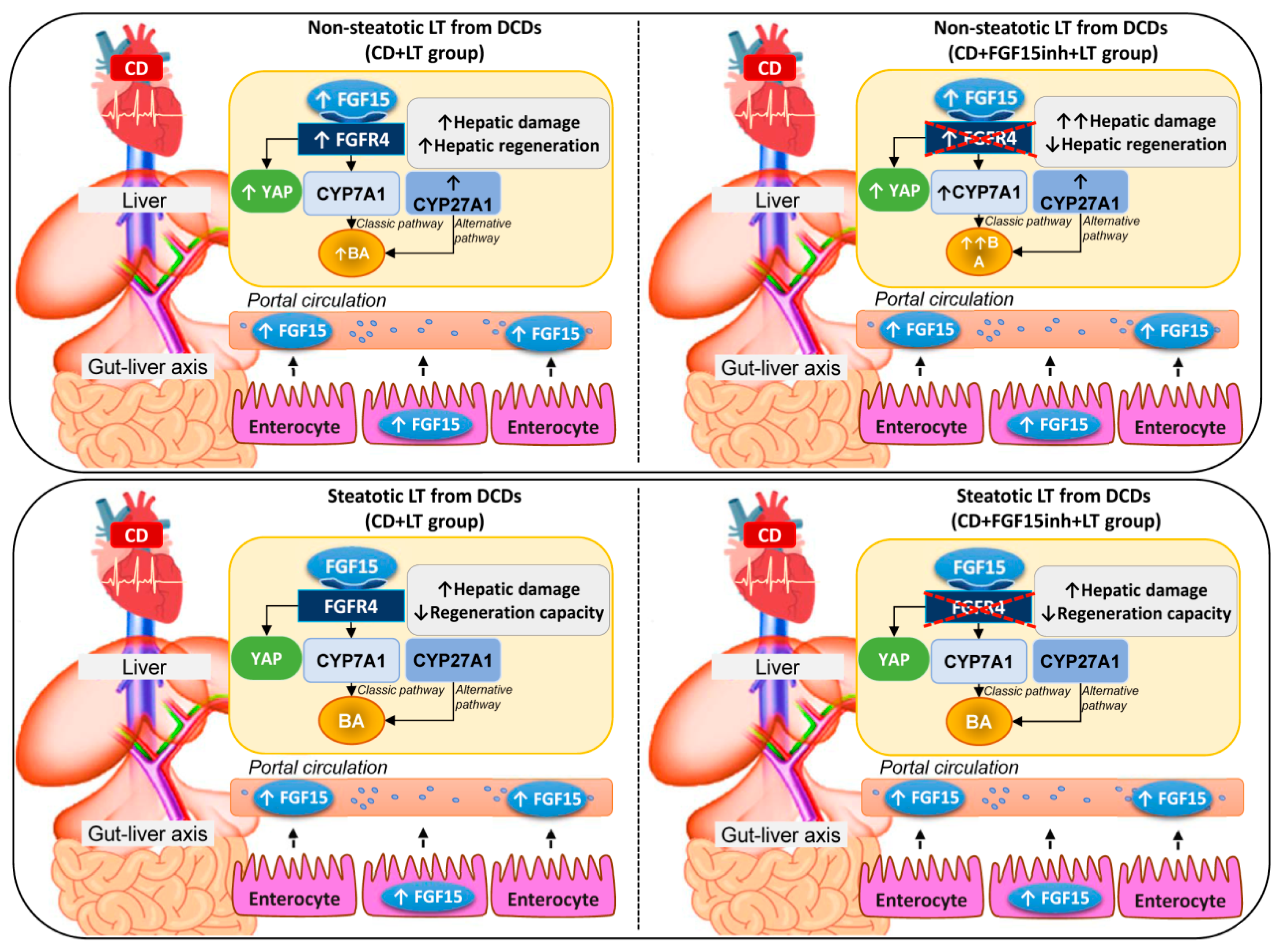
3.1. Role of FGF15 in Recipients of Non-Steatotic and Steatotic Liver Grafts from DCDs
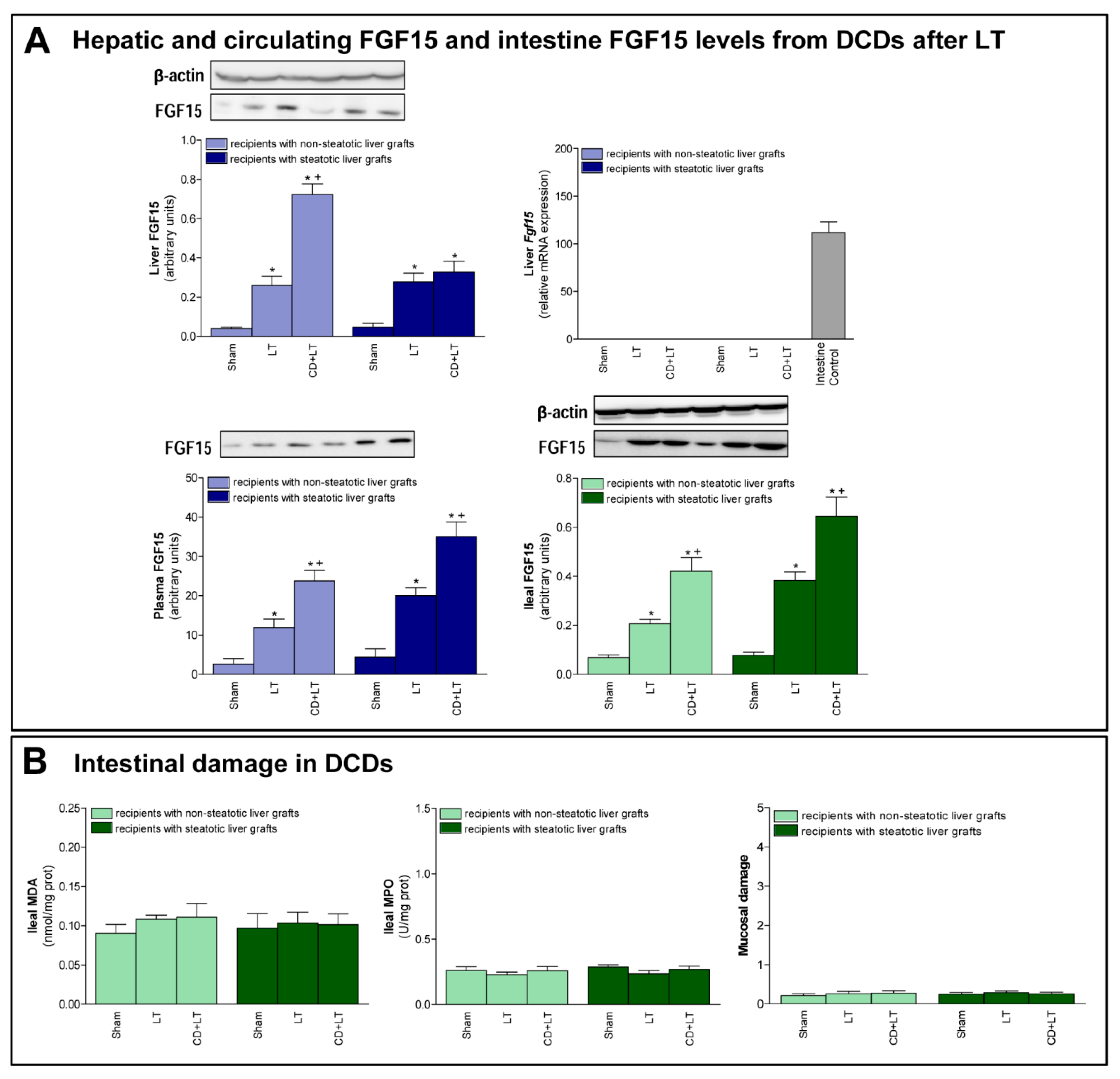
3.2. FGF15 Levels in Intestine and Plasma in LT from DCDs
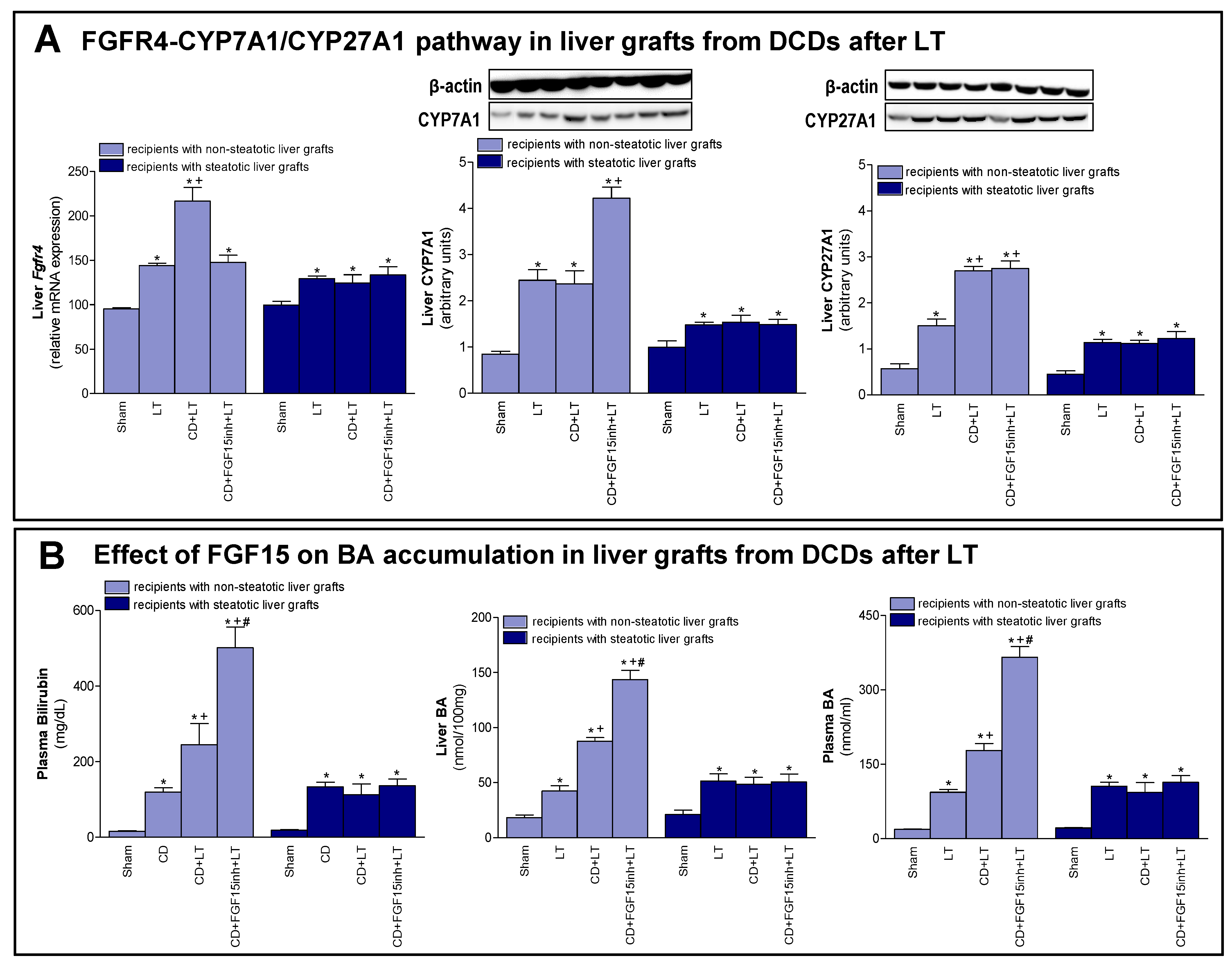
3.3. The Effects of FGF15 on the BA Synthesis Pathway in LT from DCDs
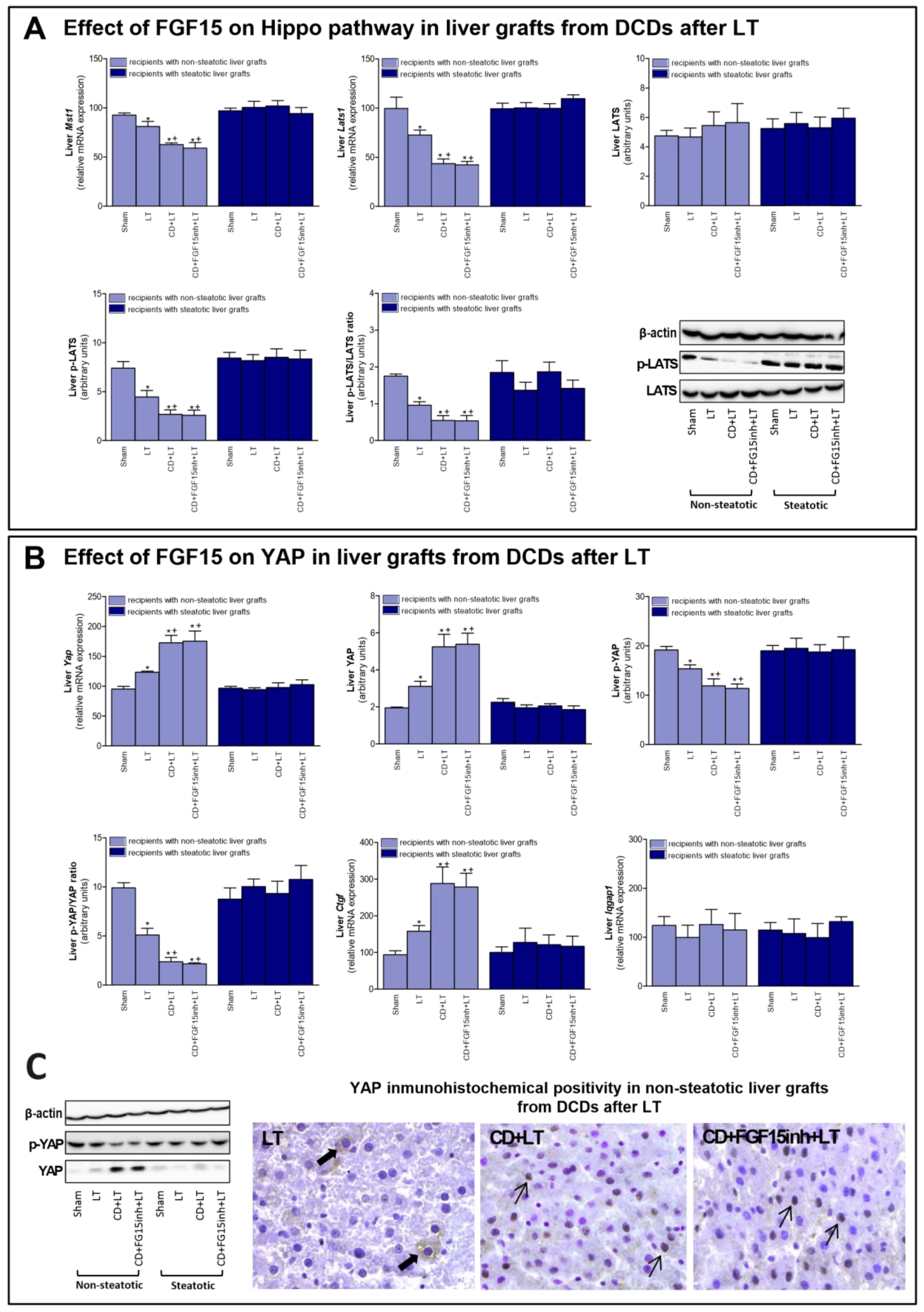
3.4. The Effect of FGF15 on the Hippo/YAP Signaling Pathway in Liver Grafts from DCDs after LT
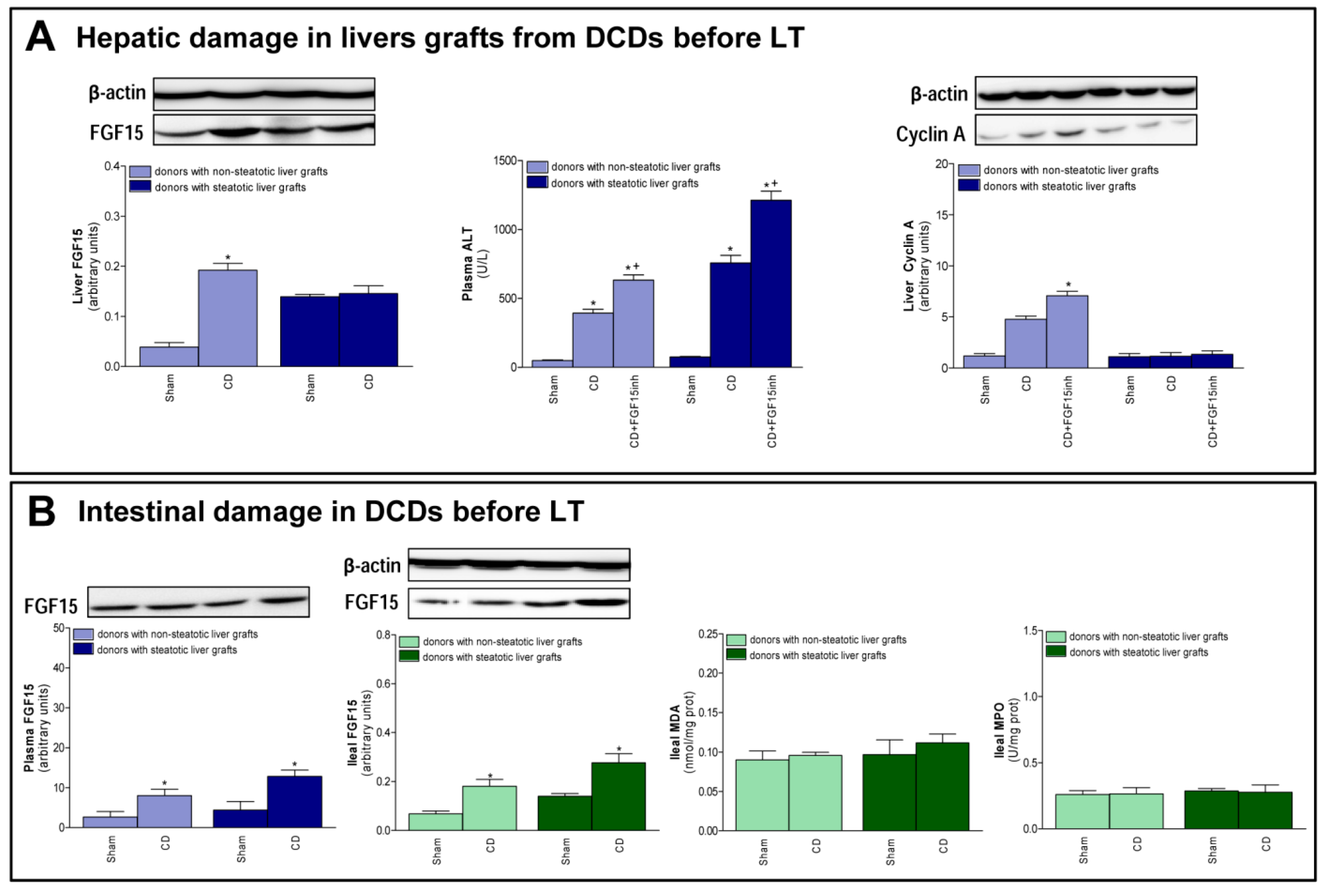
3.5. FGF15 and Related Signaling Pathways in Liver Grafts from DCDs before Procurement
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| BAs | Bile acids |
| BLC | B lymphocyte chemoattractant |
| CD | Cardiocirculatory death |
| COX-2 | Cyclooxygenase 2 |
| Ctgf | Connective tissue growth factor |
| CYP7A1 | Cytochrome P450 7A1 |
| CYP27A1 | Cytochrome P450 27A1 |
| DAB | Diaminobenzidine |
| DCDs | Donors after cardiacirculatory death |
| ERK1/2 | Extracellular signal-regulated protein kinase |
| FGF15 | Fibroblast growth factor 15 |
| FGF19 | Fibroblast growth factor 19 |
| FGFR4 | Fibroblast growth factor receptor 4 |
| G-CSF | Granulocyte colony stimulating factor |
| GM-CSF | Granulocyte macrophage colony stimulating factor |
| I/R | Ischemia-reperfusion |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IP-10 | Interferon gamma-induced protein 10 |
| IQGAP1 | IQ motif containing GTPase activating protein 1 |
| I-TAC | Interferon-inducible T-cell alpha chemoattractant |
| KC | Chemokines as keratinocyte chemoattractant |
| LATS1/2 | Large tumor suppressor 1 and 2 |
| LIX | LPS-induced CXC chemokine |
| Ln | Lean |
| LT | Liver transplantation |
| MCP | Monocyte chemoattractant protein |
| MIP | Macrophage inflammatory protein |
| MDA | Malondialdehyde |
| MPO | Myeloperoxidase |
| MST1/2 | Mammalian sterile-20 kinase 1 and 2 |
| NASH | Non-alcoholic steatohepatitis |
| Ob | Obese |
| OCT | Optimal cutting temperature |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PCNA | Proliferating cell nuclear antigen |
| PKC | Protein kinase C |
| RANTES | Regulated upon activation, normal T cell expressed and presumably secreted |
| SR-PSOX | Scavenger receptor for phosphatidylserine and oxidized low density lipoprotein |
| uPAR | Urokinase plasminogen activator receptor |
| UW | University of wisconsin |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| YAP | Yes-associated protein |
References
- Veteläinen, R.; van Vliet, A.; Gouma, D.J.; van Gulik, T.M. Steatosis as a risk factor in liver surgery. Ann. Surg 2007, 24, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Kalisvaart, M.; de Haan, J.E.; Polak, W.G.; Metselaar, H.J.; Wijnhoven, B.P.L.; Ijzermans, J.N.M.; de Jonge, J. Comparison of post-operative outcomes between donation after circulatory death and donation after brain death liver transplantation using the comprehensive complication index. Ann. Surg 2017, 266, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Goodrich, N.P.; Bragg-Gresham, J.L.; Dykstra, D.M.; Punch, J.D.; Debroy, M.A.; Greenstein, S.M.; Merion, R.M. Characteristics associated with liver graft failure: The concept of a donor risk index. Am. J. Transplant. 2006, 6, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Abt, P.; Praestgaard, J.; West, S.; Hasz, R. Donor hemodynamic profile presages graft survival in donation after cardiac death liver transplantation. Liver Transpl. 2014, 20, 165–172. [Google Scholar] [CrossRef]
- Ahmed, E.A.; El-Badry, A.M.; Hassan, A.E.A.; Redwan, A.A.; Vivarelli, M. Review on liver steatosis and its impact on liver transplantation. J. Liv Res. Dis Ther 2017, 3, 1–13. [Google Scholar] [CrossRef]
- Koneru, B.; Dikdan, G. Hepatic steatosis and liver transplantation current clinical and experimental perspectives. Transplantation 2002, 73, 325–330. [Google Scholar] [CrossRef]
- Jahn, D.; Rau, M.; Hermanns, H.M.; Geier, A. Mechanisms of enterohepatic fibroblast growth factor 15/19 signaling in health and disease. Cytokine Growth Factor Rev. 2015, 26, 625–635. [Google Scholar] [CrossRef]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.; Carotti, S.; Vespasini-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr Physiol 2013, 3, 1191–1212. [Google Scholar]
- Kong, B.; Huang, J.; Zhu, Y.; Li, G.; Williams, J.; Shen, S.; Aleksunes, L.M.; Richardson, J.R.; Apte, U.; Rudnick, D.A.; et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am. J. Physiol Gastrointest Liver Physiol 2014, 306, G893–G902. [Google Scholar] [CrossRef]
- Ji, S.; Liu, Q.; Zhang, S.; Chen, Q.; Wang, C.; Zhang, W.; Xiao, C.; Li, Y.; Nian, C.; Li, J.; et al. FGF15 activates hippo signaling to suppress bile acid metabolism and liver tumorigenesis. Dev. Cell 2019, 48, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell. Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology 2017, 152, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, P.X.; Wu, J.; Gao, Y.J.; Yin, M.X.; Lin, Y.; Yang, M.; Chen, D.P.; Sun, H.P.; Liu, Z.B.; et al. Involvement of the hippo pathway in regeneration and fibrogenesis after ischaemic acute kidney injury: YAP is the key effector. Clin. Sci. (Lond) 2016, 130, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Kakiyama, G.; Zhao, D.; Takei, H.; Fagan, A.; Hylemon, P.; Zhou, H.; Pandak, W.M.; Nittono, H.; Fiehn, O.; et al. Continued alcohol misuse in human cirrhosis is associated with an impaired gut-liver axis. Alcohol Clin Exp Res. 2017, 41, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Mutanen, A.; Lohi, J.; Heikkilä, P.; Jalanko, H.; Pakarinen, M.P. Loss of ileum decreases serum fibroblast growth factor 19 in relation to liver inflammation and fibrosis in pediatric onset intestinal failure. J. Hepatol 2015, 62, 1391–1397. [Google Scholar] [CrossRef]
- Gabbi, C.; Bertolotti, M.; Anzivino, C.; Macchioni, D.; Del Puppo, M.; Ricchi, M.; Carubbi, F.; Tagliafico, E.; Romagnoli, D.; Odoardi, M.R.; et al. Effects of bile duct ligation and cholic acid treatment on fatty liver in two rat models of non-alcoholic fatty liver disease. Dig. Liver Dis. 2012, 44, 1018–1026. [Google Scholar] [CrossRef]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef]
- Jiménez-Castro, M.B.; Meroño, N.; Mendes-Braz, M.; Gracia-Sancho, J.; Martínez-Carreres, L.; Cornide-Petronio, M.E.; Casillas-Ramirez, A.; Rodés, J.; Peralta, C. The effect of brain death in rat steatotic and non-steatotic liver transplantation with previous ischemic preconditioning. J. Hepatol. 2015, 62, 83–91. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, G.D.; Wu, L.W.; Hu, R.D. Dynamical changing patterns of histological structure and ultrastructure of liver graft undergoing warm ischemia injury from non-heart-beating donor in rats. World J. Gastroenterol. 2006, 12, 4902–4905. [Google Scholar] [CrossRef]
- Schlegel, A.; Kron, P.; Graf, R.; Dutkowski, P.; Clavien, P.A. Warm vs. cold perfusion techniques to rescue rodent liver grafts. J. Hepatol. 2014, 61, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Knoepfel, T.; Furet, P.; Mah, R.; Buschmann, N.; Leblanc, C.; Ripoche, S.; Graus-Porta, D.; Wartmann, M.; Galuba, I.; Fairhurst, R.A. 2-Formylpyridyl ureas as highly selective reversible-covalent inhibitors of fibroblast growth factor receptor 4. ACS Med. Chem Lett 2018, 9, 215–220. [Google Scholar] [CrossRef]
- Schadt, H.S.; Wolf, A.; Mahl, J.A.; Wuersch, K.; Couttet, P.; Schwald, M.; Fischer, A.; Lienard, M.; Emotte, C.; Teng, C.H.; et al. Bile acid sequestration by cholestyramine mitigates FGFR4 inhibition-induced ALT elevation. Toxicol Sci 2018, 163, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Mercado, A.I.; Negrete-Sánchez, E.; Gulfo, J.; Avalos de León, C.G.; Casillas-Ramirez, A.; Cornide-Petronio, M.E.; Bujaldon, E.; Rotondo, F.; Gracia-Sancho, J.; Jiménez-Castro, M.B.; et al. EGF-GH axis in rat steatotic and non-steatotic liver transplantation from brain-dead donors. Transplantation 2019, 103, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Rull, R.; Rimola, A.; Deulofeu, R.; Rosellò-Catafau, J.; Gelpí, E.; Rodés, J. Endogenous nitric oxide and exogenous nitric oxide supplementation in hepatic ischemia-reperfusion injury in the rat. Transplantation 2001, 71, 529–536. [Google Scholar] [CrossRef]
- Chiu, C.J.; McArdle, A.H.; Brown, R.; Scott, H.J.; Gurd, F.N. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch. Surg. 1970, 101, 478–483. [Google Scholar] [CrossRef]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology. 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Selzner, M.; Rüdiger, H.A.; Sindram, D.; Madden, J.; Clavien, P.A. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000, 32, 1280–1288. [Google Scholar] [CrossRef]
- Amersi, F.; Shen, X.D.; Moore, C.; Melinek, J.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Coito, A.J. Fibronectin-alpha 4 beta 1 integrin-mediated blockade protects genetically fat Zucker rat livers from ischemia/reperfusion injury. Am. J. Pathol. 2003, 162, 1229–1239. [Google Scholar] [CrossRef]
- Wunsch, E.; Milkiewicz, M.; Wasik, U.; Trottier, J.; Kempińska-Podhorodecka, A.; Elias, E.; Barbier, O.; Milkiewicz, P. Expression of hepatic fibroblast growth factor 19 is enhanced in primary biliary cirrhosis and correlates with severity of the disease. Sci Rep. 2015, 5, 13462. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; van der Gaag, N.A.; Gouma, D.J.; Jansen, P.L. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology 2009, 49, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Tarlow, B.D.; Fedorov, L.M.; Taylor, M.; Pelz, C.; Li, B.; Darnell, J.; Grompe, M. Fibroblast Growth Factor Signaling Controls Liver Size in Mice With Humanized Livers. Gastroenterology 2015, 149, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Anakk, S.; Bhosale, M.; Schmidt, V.A.; Johnson, R.L.; Finegold, M.J.; Moore, D.D. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013, 5, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, I.V.; Yarygin, K.N. Cellular mechanisms of liver regeneration and cell-based therapies of liver diseases. Biomed. Res. Int. 2017, 2017, 8910821. [Google Scholar] [CrossRef]
- Desdouets, C.; Thoresen, G.H.; Senamaud-Beaufort, C.; Christoffersen, T.; Brechot, C.; Sobczak-Thepot, J. cAMP-dependent positive control of cyclin A2 expression during G1/S transition in primary hepatocytes. Biochem Biophys Res. Commun 1999, 261, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Mörk, L.M.; Li, M.; Sandblom, A.L.; Björkhem, I.; Höijer, J.; Ericzon, B.G.; Jorns, C.; Gilg, S.; Sparrelid, E.; et al. Circulating fibroblast growth factor 19 in portal and systemic blood. J. Clin Exp Hepatol 2018, 8, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Fernandez-Barrena, M.G.; Urtasun, R.; Elizalde, M.; Barcena-Varela, M.; Jiménez, M.; Chang, H.C.; Barbero, R.; et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: Development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut. 2017, 66, 1818–1828. [Google Scholar] [CrossRef]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun. 2017, 1, 1024–1042. [Google Scholar] [CrossRef]
- Varela-Rey, M.; Embade, N.; Ariz, U.; Lu, S.C.; Mato, J.M.; Martínez-Chantar, M.L. Non-alcoholic steatohepatitis and animal models: Understanding the human disease. Int. J. Biochem. Cell Biol. 2009, 41, 969–976. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal models of nonalcoholic fatty liver disease-A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory links between high fat diets and diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Castro, M.B.; Casillas-Ramírez, A.; Mendes-Braz, M.; Massip-Salcedo, M.; Gracia-Sancho, J.; Elias-Miró, M.; Rodés, J.; Peralta, C. Adiponectin and resistin protect steatotic livers undergoing transplantation. J. Hepatol. 2013, 59, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Elias-Miró, M.; Mendes-Braz, M.; Cereijo, R.; Villarroya, F.; Jiménez-Castro, M.B.; Gracia-Sancho, J.; Guixé-Muntet, S.; Massip-Salcedo, M.; Domingo, J.C.; Bermudo, R.; et al. Resistin and visfatin in steatotic and non-steatotic livers in the setting of partial hepatectomy under ischemia-reperfusion. J. Hepatol. 2014, 60, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Alfany-Fernandez, I.; Casillas-Ramirez, A.; Bintanel-Morcillo, M.; Brosnihan, K.B.; Ferrario, C.M.; Serafin, A.; Rimola, A.; Rodés, J.; Roselló-Catafau, J.; Peralta, C. Therapeutic targets in liver transplantation: Angiotensin II in nonsteatotic grafts and angiotensin-(1-7) in steatotic grafts. Am. J. Transplant. 2009, 9, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, F.S.; Alfany-Fernandez, I.; Casillas-Ramirez, A.; Massip-Salcedo, M.; Serafín, A.; Rimola, A.; Arroyo, V.; Rodés, J.; Roselló-Catafau, J.; Peralta, C. Are angiotensin II receptor antagonists useful strategies in steatotic and nonsteatotic livers in conditions of partial hepatectomy under ischemia-reperfusion? J. Pharmacol Exp. Ther. 2009, 329, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Jimenez, M.; Barcena-Varela, M.; Santamaría, E.; Urtasun, R.; Rodriguez-Ortigosa, C.; Prieto, J.; Berraondo, P.; et al. Bile acids, FGF15/19 and liver regeneration: From mechanisms to clinical applications. Biochim Biophys Acta Mol Basis Dis 2018, 1864, 1326–1334. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Zhang, F.; Yu, L.; Lin, X.; Cheng, P.; He, L.; Li, X.; Lu, X.; Tan, Y.; Yang, H.; Cai, L.; et al. Minireview: Roles of fibroblast growth factors 19 and 21 in metabolic regulation and chronic diseases. Mol. Endocrinol 2015, 29, 1400–1413. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Vaz, F.M.; Ferdinandusse, S. Bile acid analysis in human disorders of bile acid biosynthesis. Mol Asp. Med. 2017, 56, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yin, P.; Tang, L.; Xing, W.; Huang, Q.; Cao, D.; Zhao, X.; Wang, W.; Lu, X.; Xu, Z.; et al. Metabolomics study of stepwise hepatocarcinogenesis from the model rats to patients: Potential biomarkers effective for small hepatocellular carcinoma diagnosis. Mol Cell Proteomics. 2012, 11, M111.010694. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.F.; Varghese, R.S.; Zhou, B.; Nezami Ranjbar, M.R.; Zhao, Y.; Tsai, T.H.; Di Poto, C.; Wang, J.; Goerlitz, D.; Luo, Y.; et al. LC-MS based serum metabolomics for identification of hepatocellular carcinoma biomarkers in Egyptian cohort. J. Proteome Res. 2012, 11, 5914–5923. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.; Jaeschke, H.; Copple, B.L. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 2011, 178, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Jiménez-Castro, M.B.; Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: Effects on the liver sinusoidal milieu. J. Hepatol. 2013, 59, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: A 2015 update. Clin Sci (Lond). 2015, 129, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.; Kim, N.D.; Moon, J.O.; Copple, B.L. Upregulation of early growth response factor-1 by bile acids requires mitogen-activated protein kinase signaling. Toxicol Appl. Pharmacol. 2010, 243, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science. 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Stravitz, R.T.; Dent, P.; Hylemon, P.B. Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J. Biol Chem. 2001, 276, 15816–15822. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.P.; Stravitz, R.T.; Vlahcevic, Z.R.; Gurley, E.C.; Sando, J.J.; Hylemon, P.B. Activation of protein kinase C alpha and delta by bile acids: Correlation with bile acid structure and diacylglycerol formation. J. Lipid Res. 1997, 38, 2446–2454. [Google Scholar]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA. 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.K.; Graf, D.; Schmitt, M.; Vom Dahl, S.; Haussinger, D. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology. 2001, 121, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kim, S.E.; Lee, Y.S. SP600125, a selective JNK inhibitor, aggravates hepatic ischemia-reperfusion injury. Exp Mol Med. 2006, 38, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Massip-Salcedo, M.; Casillas-Ramirez, A.; Franco-Gou, R.; Bartrons, R.; Ben Mosbah, I.; Serafin, A.; Rosello-Catafau, J.; Peralta, C. Heat shock proteins and mitogen-activaed protein kinases in steatotic livers undergoing ischemia-reperfusion: Some answers. Am. J. Pathol. 2006, 168, 1474–1485. [Google Scholar] [CrossRef]
- Yuan, W.C.; Pepe-Mooney, B.; Galli, G.G.; Dill, M.T.; Huang, H.T.; Hao, M.; Wang, Y.; Liang, H.; Calogero, R.A.; Camargo, F.D. NUAK2 is a critical YAP target in liver cancer. Nat. Commun 2018, 9, 4834. [Google Scholar] [CrossRef]
- Sayedyahossein, S.; Li, Z.; Hedman, A.C.; Morgan, C.J.; Sacks, D.B. IQGAP1 binds to yes-associated protein (YAP) and modulates its transcriptional activity. J. Biol. Chem. 2016, 291, 19261–19273. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y. Role of FGF19 induced FGFR4 activation in the regulation of glucose homeostasis. Aging (Albany NY) 2009, 1, 1023–1027. [Google Scholar] [CrossRef]
- Araújo, T.G.; de Oliveira, A.G.; Tobar, N.; Saad, M.J.; Moreira, L.R.; Reis, E.R.; Nicola, E.M.; de Jorge, G.L.; dos Tártaro, R.R.; Boin, I.F.; et al. Liver regeneration following partial hepatectomy is improved by enhancing the HGF/Met axis and Akt and Erk pathways after low-power laser irradiation in rats. Lasers Med. Sci. 2013, 28, 1511–1517. [Google Scholar] [CrossRef]
- Murata, S.; Maruyama, T.; Nowatari, T.; Takahashi, K.; Ohkohchi, N. Signal transduction of platelet-induced liver regeneration and decrease of liver fibrosis. Int. J. Mol. Sci. 2014, 15, 5412–5425. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Y.W.; Yao, A.H.; Yu, Y.; Hua, Z.Y.; Pu, L.Y.; Li, G.Q.; Li, X.C.; Zhang, F.; Sheng, G.Q.; et al. Adenoviral cardiotrophin-1 transfer improves survival and early graft function after ischemia and reperfusion in rat small-for-size liver transplantation model. Transpl. Int. 2008, 21, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Ben Mosbah, I.; Alfany-Fernandez, I.; Martel, C.; Zaouali, M.A.; Bintanel-Morcillo, M.; Rimola, A.; Rodes, J.; Brenner, C.; Rosello-Catafau, J.; Peralta, C. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010, 1, e52. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, M.; Chen, S.W.; Brown, K.M.; D’Agati, V.D.; Lee, H.T. Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P1 receptor activation. Lab. Investig. 2010, 90, 1209–1224. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avalos-de León, C.G.; Jiménez-Castro, M.B.; Cornide-Petronio, M.E.; Gulfo, J.; Rotondo, F.; Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. The Effect of Fibroblast Growth Factor 15 Signaling in Non-Steatotic and Steatotic Liver Transplantation from Cardiocirculatory Death. Cells 2019, 8, 1640. https://doi.org/10.3390/cells8121640
Avalos-de León CG, Jiménez-Castro MB, Cornide-Petronio ME, Gulfo J, Rotondo F, Gracia-Sancho J, Casillas-Ramírez A, Peralta C. The Effect of Fibroblast Growth Factor 15 Signaling in Non-Steatotic and Steatotic Liver Transplantation from Cardiocirculatory Death. Cells. 2019; 8(12):1640. https://doi.org/10.3390/cells8121640
Chicago/Turabian StyleAvalos-de León, Cindy G., Mónica B. Jiménez-Castro, María Eugenia Cornide-Petronio, José Gulfo, Floriana Rotondo, Jordi Gracia-Sancho, Araní Casillas-Ramírez, and Carmen Peralta. 2019. "The Effect of Fibroblast Growth Factor 15 Signaling in Non-Steatotic and Steatotic Liver Transplantation from Cardiocirculatory Death" Cells 8, no. 12: 1640. https://doi.org/10.3390/cells8121640
APA StyleAvalos-de León, C. G., Jiménez-Castro, M. B., Cornide-Petronio, M. E., Gulfo, J., Rotondo, F., Gracia-Sancho, J., Casillas-Ramírez, A., & Peralta, C. (2019). The Effect of Fibroblast Growth Factor 15 Signaling in Non-Steatotic and Steatotic Liver Transplantation from Cardiocirculatory Death. Cells, 8(12), 1640. https://doi.org/10.3390/cells8121640