Post-ER Stress Biogenesis of Golgi Is Governed by Giantin


 , , , and
, , , and 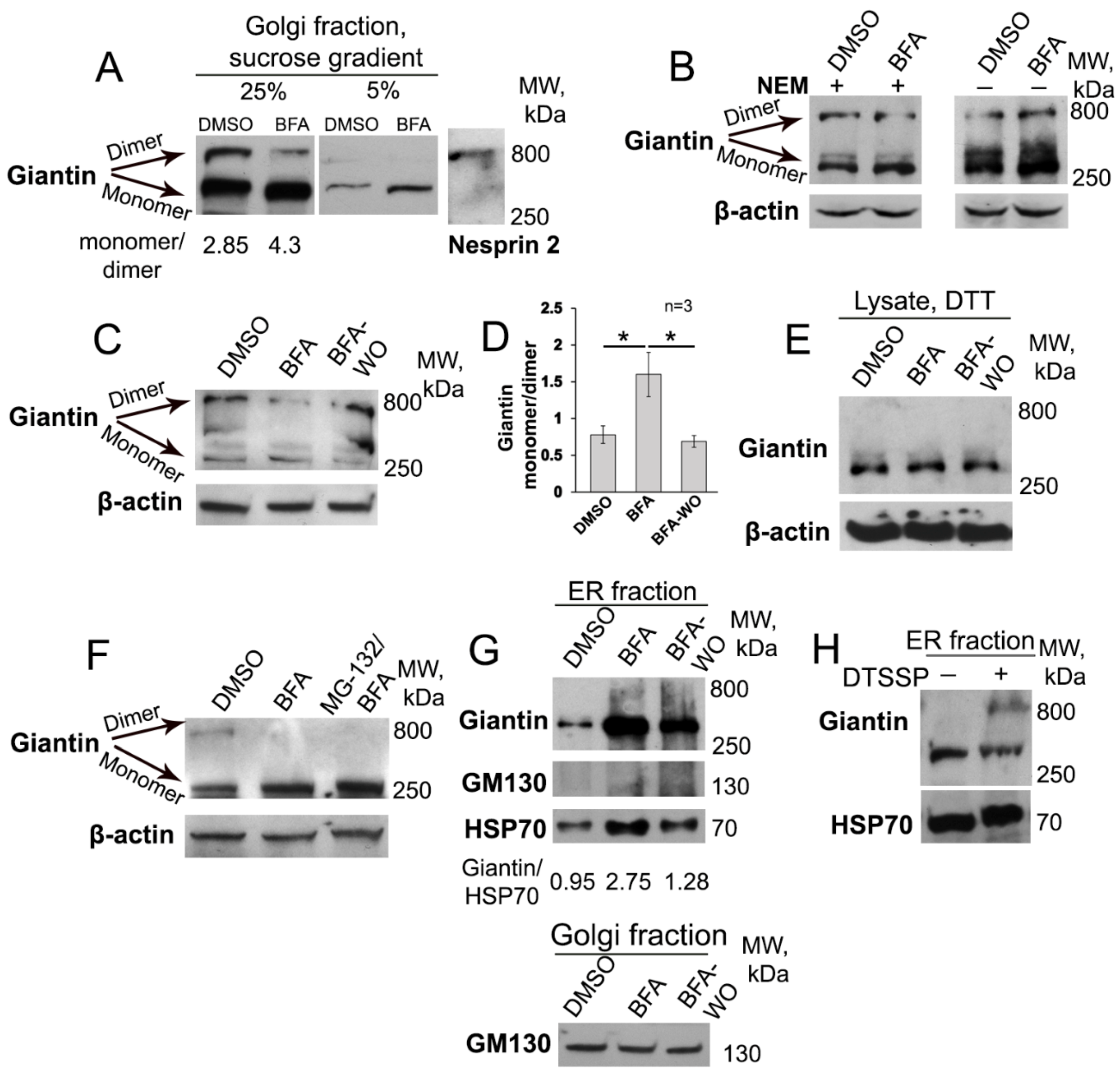{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Antibodies and Reagents
2.2. Immunoprecipitation (IP), Plasmid Constructions and Transfection
2.3. In Vitro Crosslinking
2.4. Confocal Immunofluorescence Microscopy
2.5. Three-Dimensional Structured Illumination (3D-SIM) Microscopy and Image Analysis
2.6. AFM Imaging and Image Analysis
2.7. In Situ Proximity Ligation Assay
2.8. Isolation of Golgi Membrane Fractions by Sucrose Gradient Centrifugation
2.9. Zonal Sedimentation Analysis on Sucrose Gradients
2.10. Isolation of Microsomal Fraction
2.11. Statistical Analysis
2.12. Miscellaneous
3. Results
3.1. BFA-induced Golgi Disorganization Is Associated with Giantin Monomerization
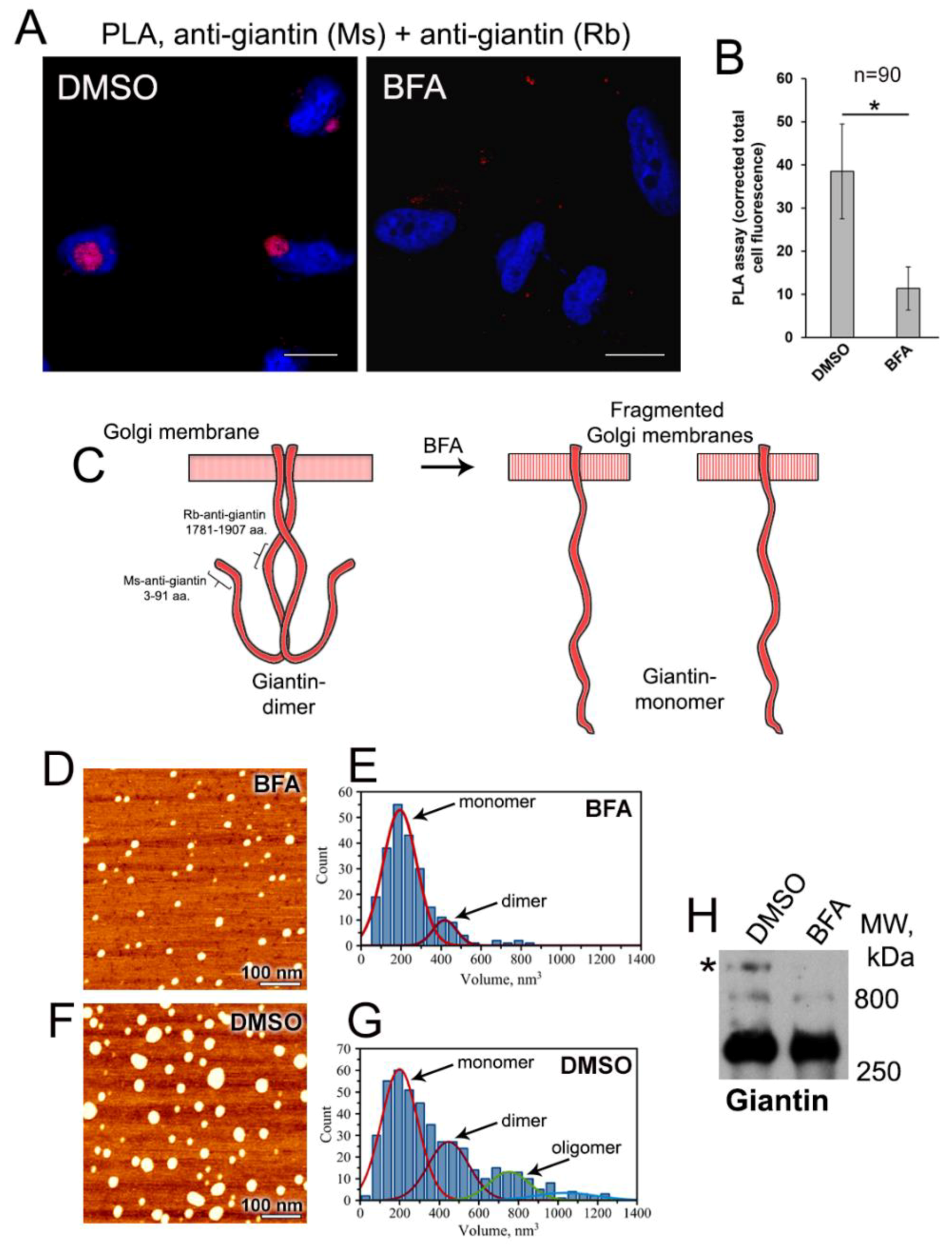
3.2. Examination of Giantin-Giantin Interaction by Proximity Ligation Assay
3.3. AFM Characterization of Giantin Protein
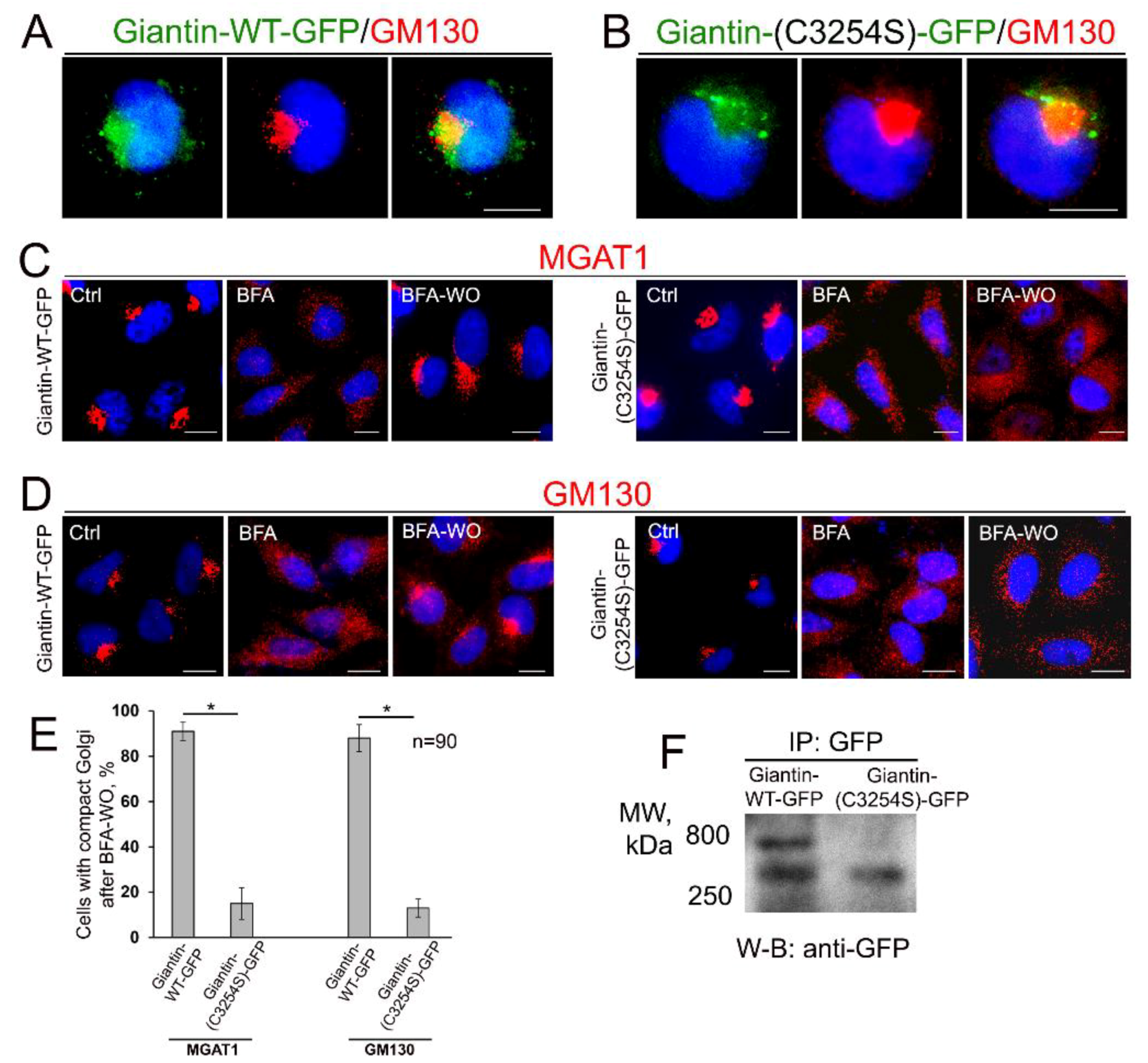
3.4. Giantin Dimerization Is Critical for Golgi Biogenesis
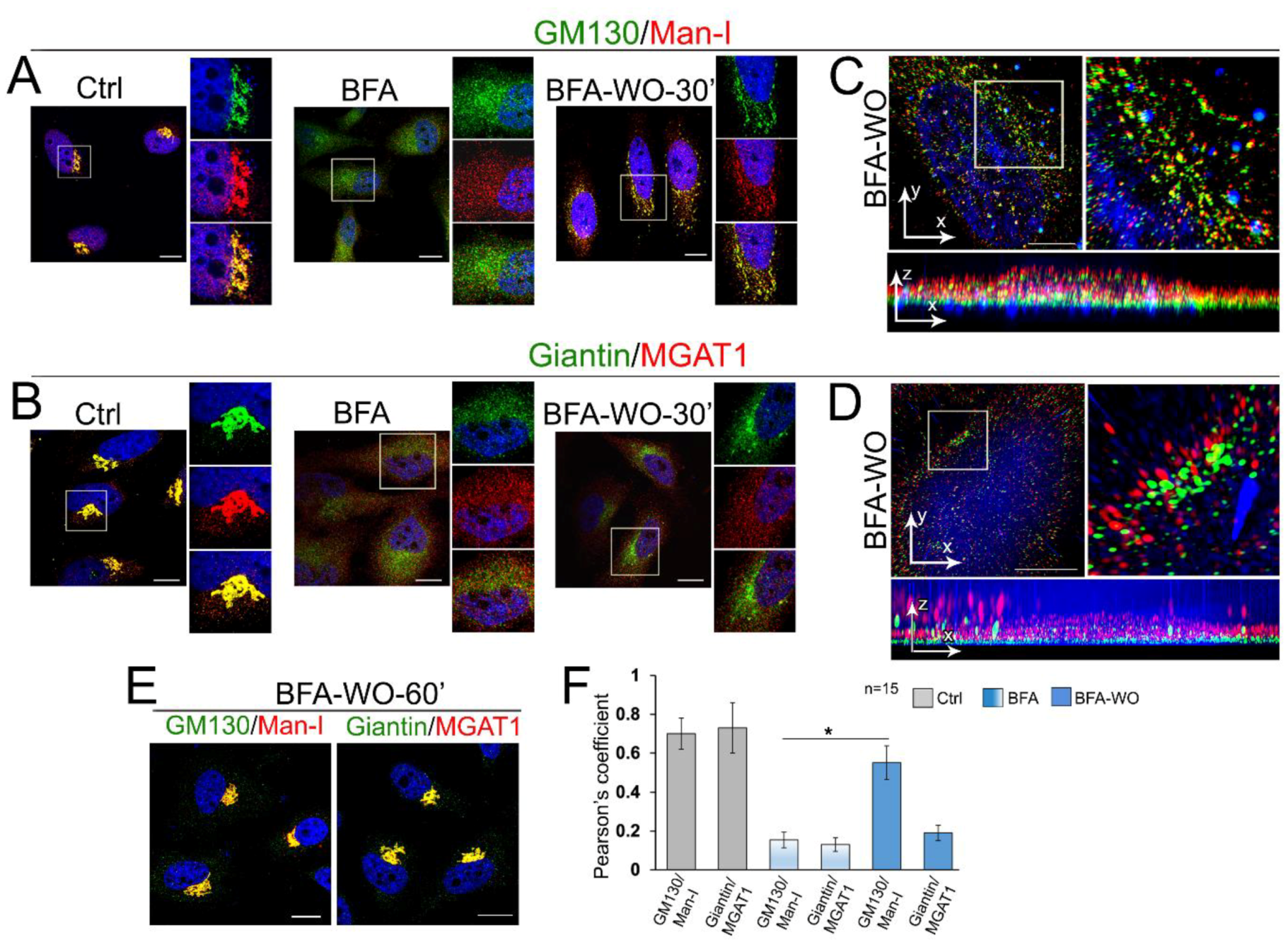
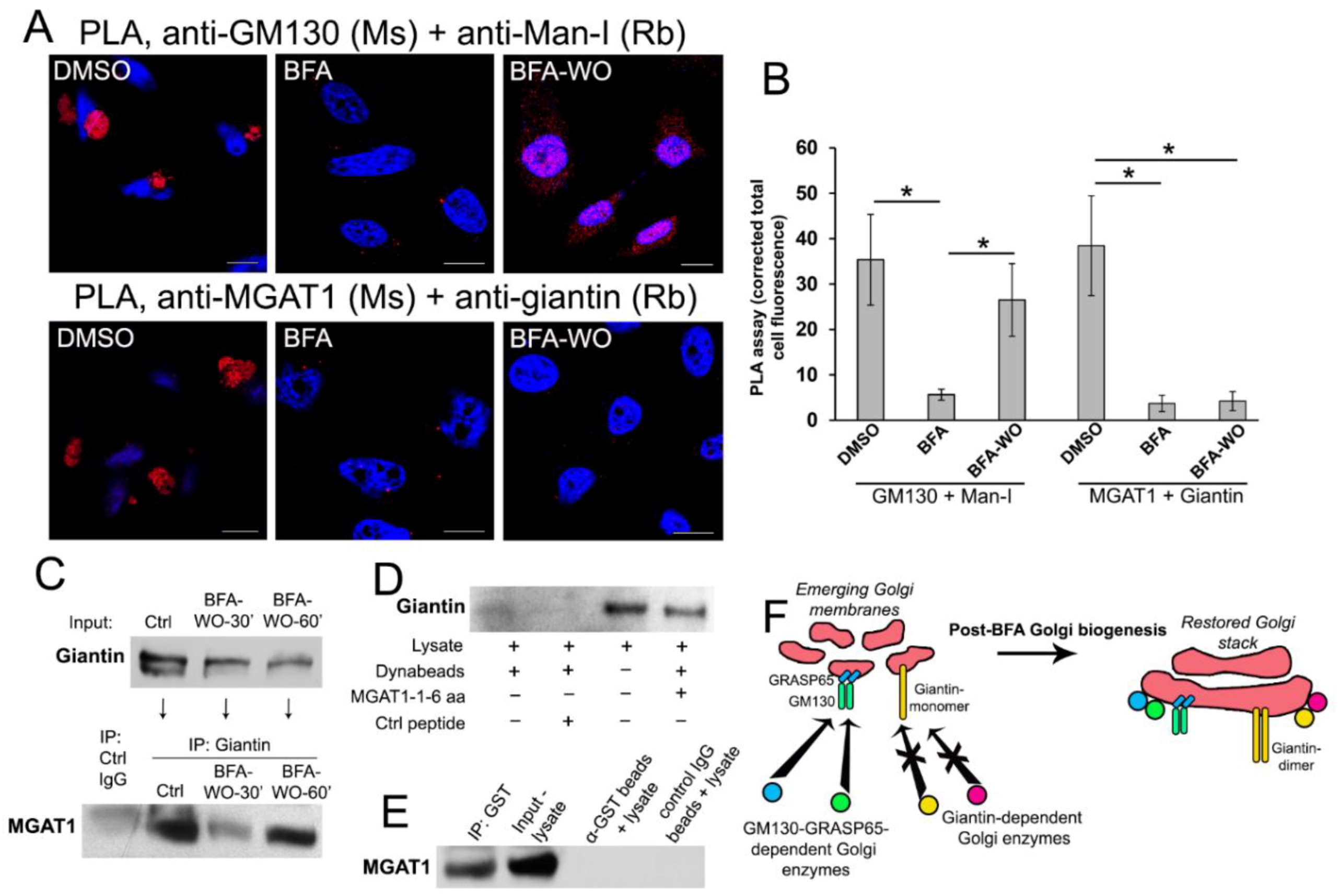
3.5. Giantin Dictates Sequential Targeting of Golgi Residential Enzymes in BFA Washout Cells
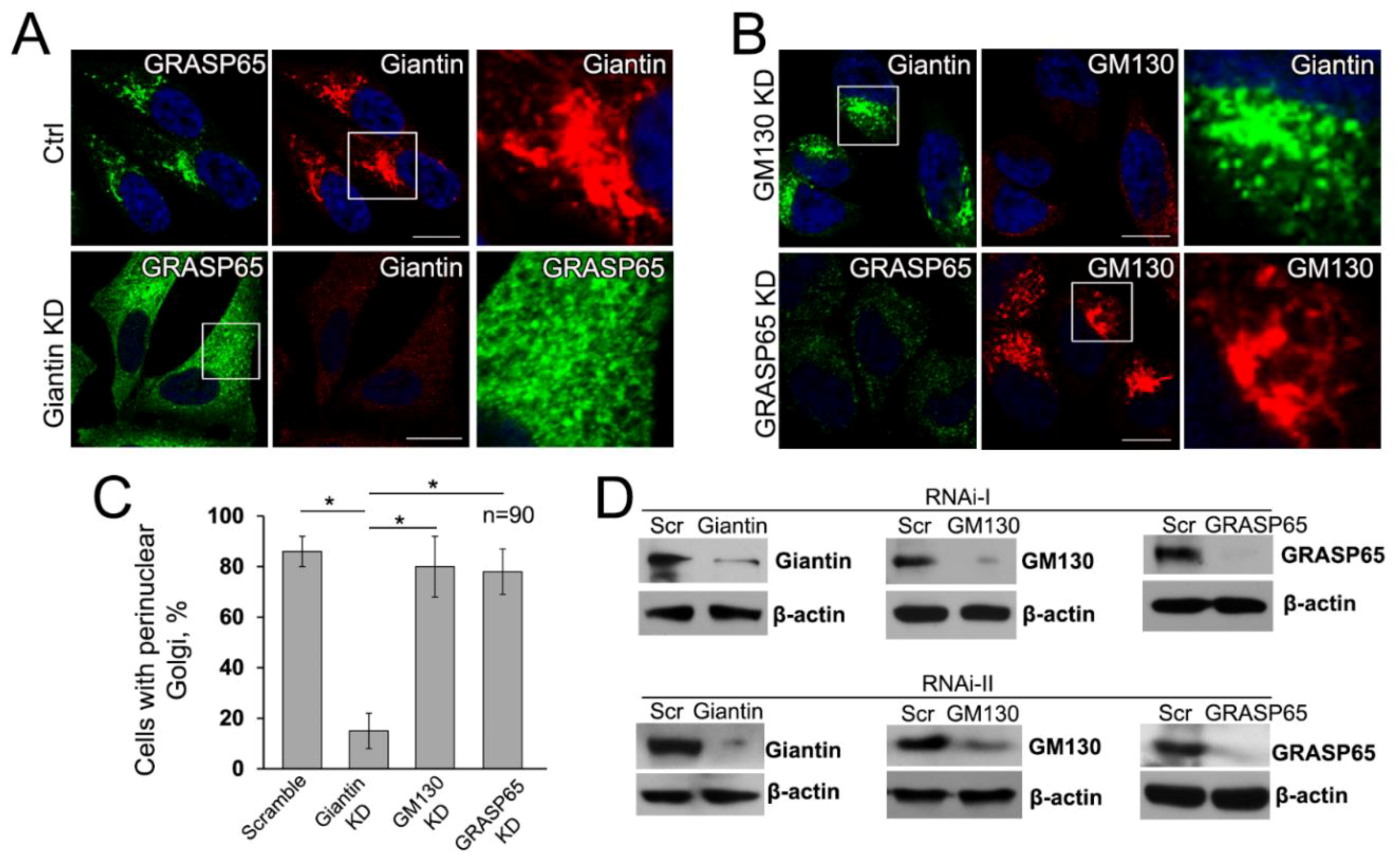
3.6. Giantin, but not Other Golgi Proteins, Is the Main Driver of Golgi Biogenesis
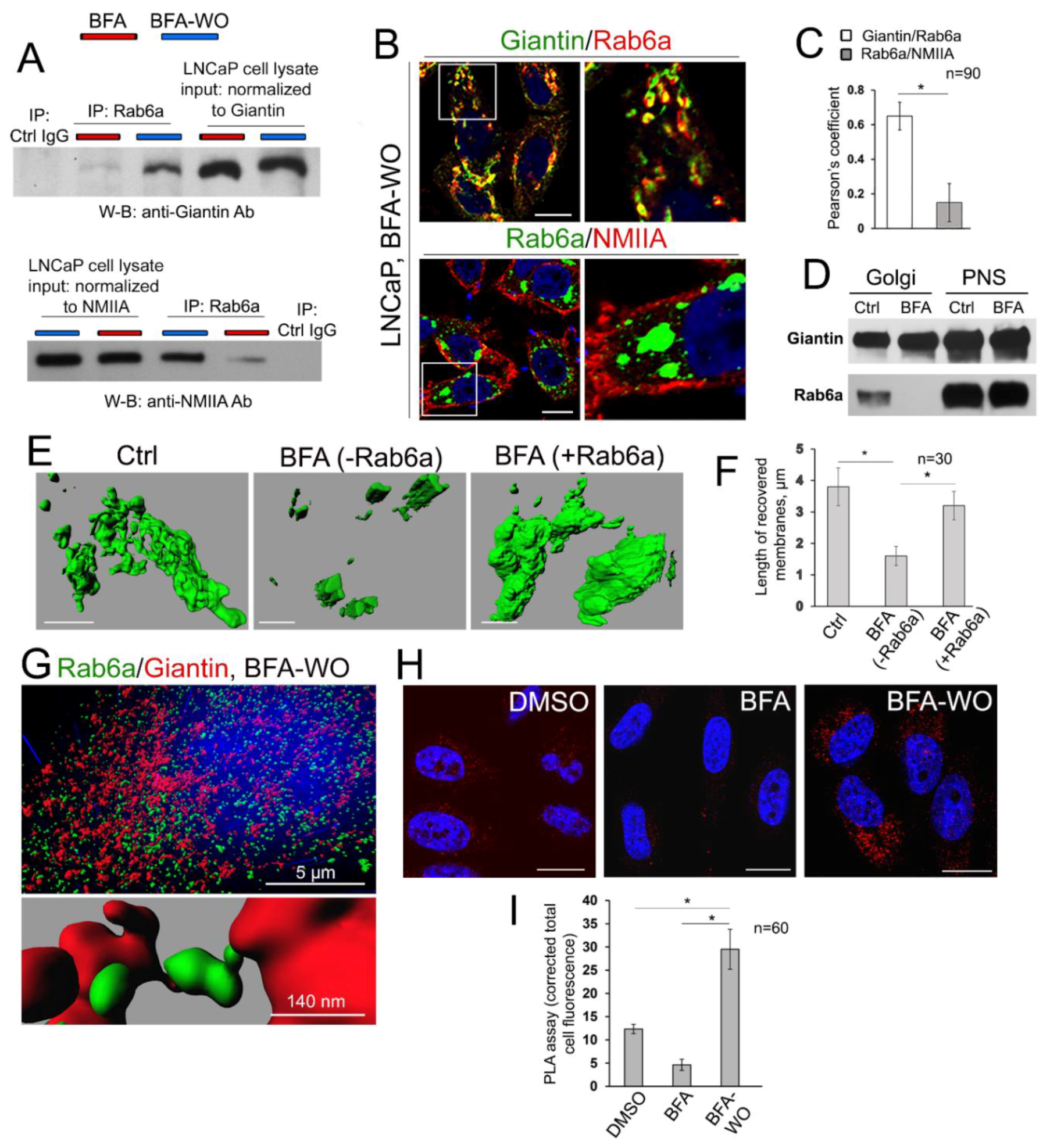
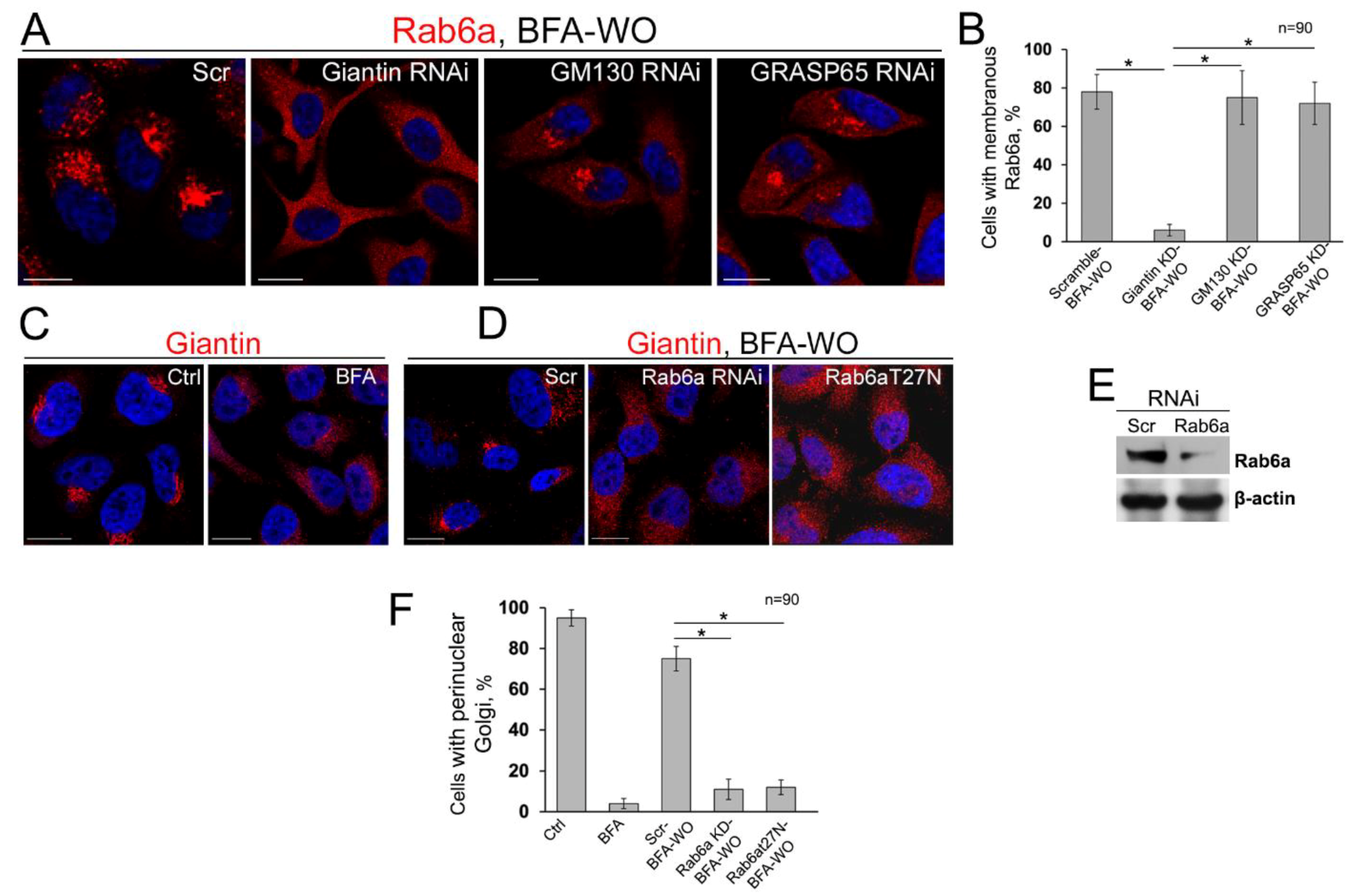
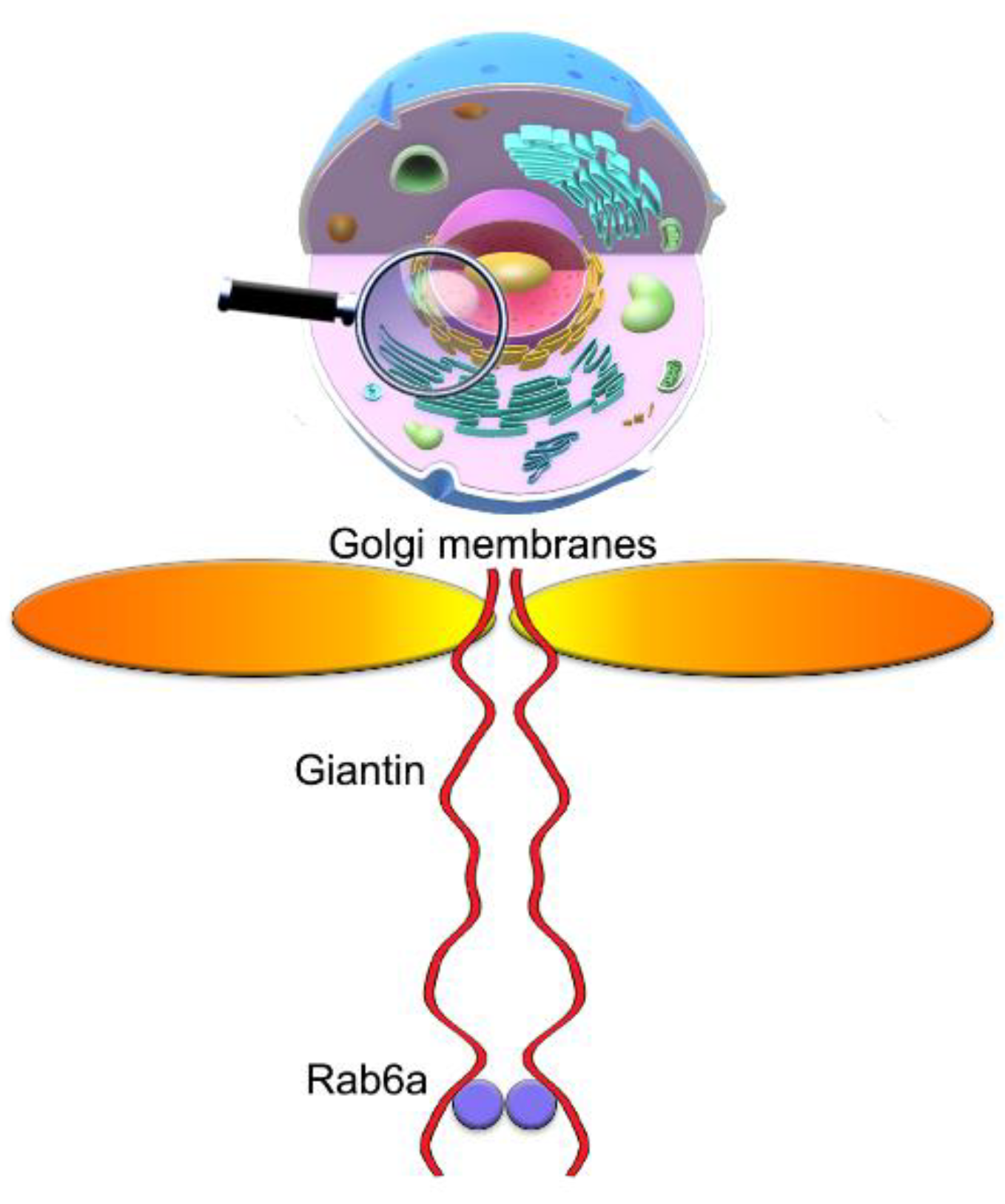
3.7. The Mutuality of Rab6a and Giantin
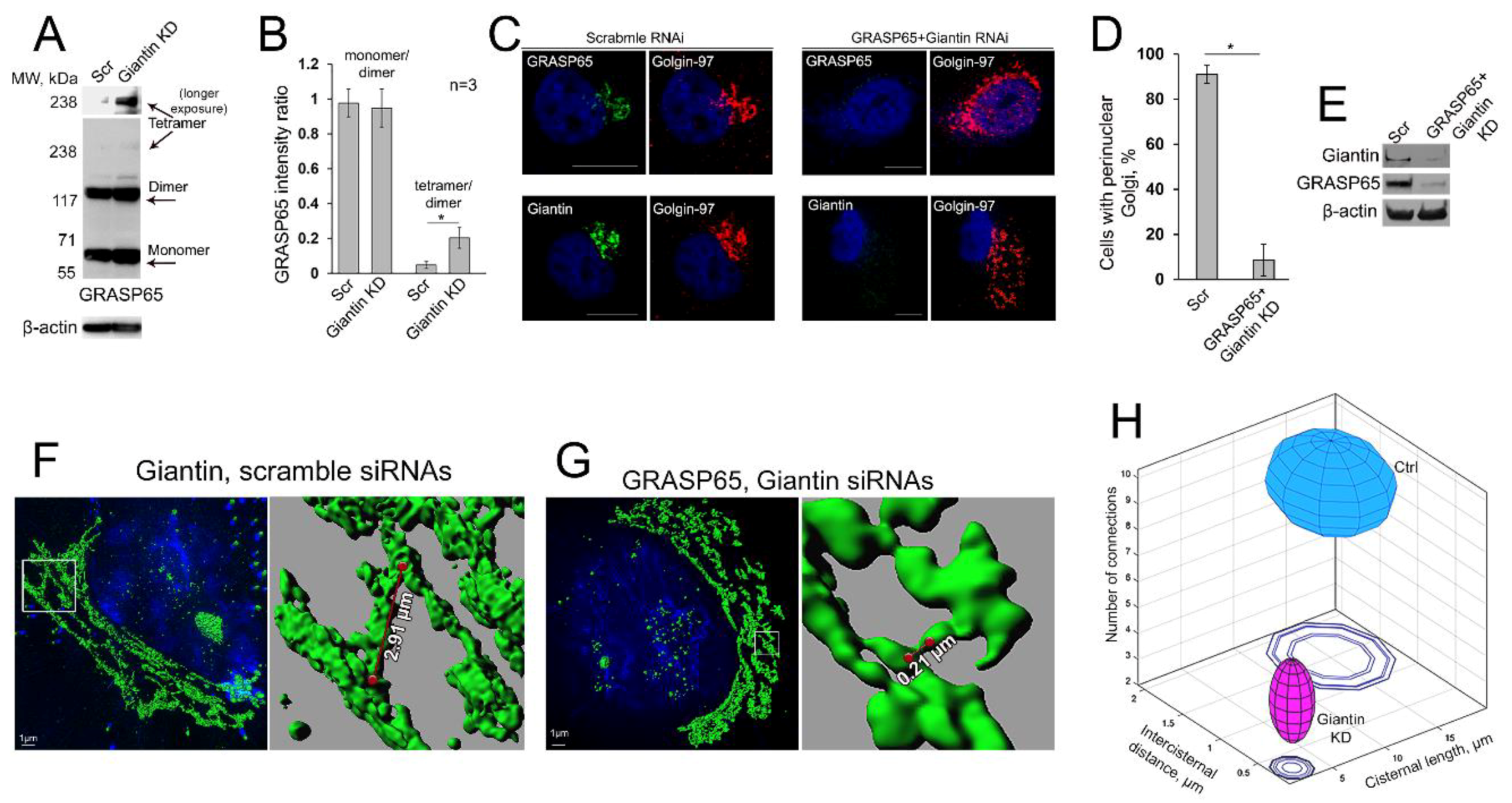
3.8. Is GRASP65 the Protein that May Compensate for the Lack of Giantin?
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pulvirenti, T.; Giannotta, M.; Capestrano, M.; Capitani, M.; Pisanu, A.; Polishchuk, R.S.; San Pietro, E.; Beznoussenko, G.V.; Mironov, A.A.; Turacchio, G.; et al. A traffic-activated golgi-based signalling circuit coordinates the secretory pathway. Nat. Cell Biol. 2008, 10, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Rothman, J.E. The future of golgi research. Mol. Biol. Cell 2010, 21, 3776–3780. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Hui, N.; Hunte, F.; Kieckbusch, R.; Berger, E.G.; Warren, G.; Nilsson, T. Mapping the distribution of golgi enzymes involved in the construction of complex oligosaccharides. J. Cell Sci. 1995, 108, 1617–1627. [Google Scholar] [PubMed]
- Nilsson, T.; Au, C.E.; Bergeron, J.J. Sorting out glycosylation enzymes in the golgi apparatus. FEBS Lett. 2009, 583, 3764–3769. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.; Stamnes, M.; Ravazzola, M.; Amherdt, M.; Perrelet, A.; Sollner, T.H.; Rothman, J.E. Bidirectional transport by distinct populations of copi-coated vesicles. Cell 1997, 90, 335–349. [Google Scholar] [CrossRef]
- Mironov, A.A.; Weidman, P.; Luini, A. Variations on the intracellular transport theme: Maturing cisternae and trafficking tubules. J. Cell Biol. 1997, 138, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Zaal, K.J.; Smith, C.L.; Polishchuk, R.S.; Altan, N.; Cole, N.B.; Ellenberg, J.; Hirschberg, K.; Presley, J.F.; Roberts, T.H.; Siggia, E.; et al. Golgi membranes are absorbed into and reemerge from the er during mitosis. Cell 1999, 99, 589–601. [Google Scholar] [CrossRef]
- Petrosyan, A.; Cheng, P.W. Golgi fragmentation induced by heat shock or inhibition of heat shock proteins is mediated by non-muscle myosin iia via its interaction with glycosyltransferases. Cell Stress Chaperones 2014, 19, 241–254. [Google Scholar] [CrossRef]
- Pavelka, M.; Ellinger, A. Effect of colchicine on the golgi complex of rat pancreatic acinar cells. J. Cell Biol. 1983, 97, 737–748. [Google Scholar] [CrossRef]
- Rogalski, A.A.; Bergmann, J.E.; Singer, S.J. Effect of microtubule assembly status on the intracellular processing and surface expression of an integral protein of the plasma membrane. J. Cell Biol. 1984, 99, 1101–1109. [Google Scholar] [CrossRef]
- Klausner, R.D.; Donaldson, J.G.; Lippincott-Schwartz, J. Brefeldin a: Insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992, 116, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.M.; Renau-Piqueras, J.; Marin, M.P.; Esteban-Pretel, G. Chronic alcohol exposure affects the cell components involved in membrane traffic in neuronal dendrites. Neurotox. Res. 2015, 27, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A.; Cheng, P.W.; Clemens, D.L.; Casey, C.A. Downregulation of the small gtpase sar1a: A key event underlying alcohol-induced golgi fragmentation in hepatocytes. Sci. Rep. 2015, 5, 17127. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. The concept of self-organization in cellular architecture. J. Cell Biol. 2001, 155, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Linstedt, A.D. Capacity of the golgi apparatus for biogenesis from the endoplasmic reticulum. Mol. Biol. Cell 2003, 14, 5011–5018. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid redistribution of golgi proteins into the er in cells treated with brefeldin a: Evidence for membrane cycling from golgi to er. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Berger, E.G.; Grimm, K.; Bachi, T.; Bosshart, H.; Kleene, R.; Watzele, M. Double immunofluorescent staining of alpha 2,6 sialyltransferase and beta 1,4 galactosyltransferase in monensin-treated cells: Evidence for different golgi compartments? J. Cell Biochem. 1993, 52, 275–288. [Google Scholar] [CrossRef]
- Sweeney, D.A.; Siddhanta, A.; Shields, D. Fragmentation and re-assembly of the golgi apparatus in vitro. A requirement for phosphatidic acid and phosphatidylinositol 4,5-bisphosphate synthesis. J. Biol. Chem. 2002, 277, 3030–3039. [Google Scholar] [CrossRef]
- Zilberman, Y.; Alieva, N.O.; Miserey-Lenkei, S.; Lichtenstein, A.; Kam, Z.; Sabanay, H.; Bershadsky, A. Involvement of the rho-mdia1 pathway in the regulation of golgi complex architecture and dynamics. Mol. Biol. Cell 2011, 22, 2900–2911. [Google Scholar] [CrossRef]
- Misumi, Y.; Misumi, Y.; Miki, K.; Takatsuki, A.; Tamura, G.; Ikehara, Y. Novel blockade by brefeldin a of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 1986, 261, 11398–11403. [Google Scholar]
- Peyroche, A.; Antonny, B.; Robineau, S.; Acker, J.; Cherfils, J.; Jackson, C.L. Brefeldin a acts to stabilize an abortive arf-gdp-sec7 domain protein complex: Involvement of specific residues of the sec7 domain. Mol. Cell 1999, 3, 275–285. [Google Scholar] [CrossRef]
- Mossessova, E.; Corpina, R.A.; Goldberg, J. Crystal structure of arf1*sec7 complexed with brefeldin a and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 2003, 12, 1403–1411. [Google Scholar] [CrossRef]
- Renault, L.; Guibert, B.; Cherfils, J. Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 2003, 426, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Kreis, T.E.; Lowe, M.; Pepperkok, R. Cops regulating membrane traffic. Annu. Rev. Cell Dev. Biol. 1995, 11, 677–706. [Google Scholar] [CrossRef] [PubMed]
- Sciaky, N.; Presley, J.; Smith, C.; Zaal, K.J.; Cole, N.; Moreira, J.E.; Terasaki, M.; Siggia, E.; Lippincott-Schwartz, J. Golgi tubule traffic and the effects of brefeldin a visualized in living cells. J. Cell Biol. 1997, 139, 1137–1155. [Google Scholar] [CrossRef]
- Guo, Y.; Punj, V.; Sengupta, D.; Linstedt, A.D. Coat-tether interaction in golgi organization. Mol. Biol. Cell 2008, 19, 2830–2843. [Google Scholar] [CrossRef]
- Myhill, N.; Lynes, E.M.; Nanji, J.A.; Blagoveshchenskaya, A.D.; Fei, H.; Carmine Simmen, K.; Cooper, T.J.; Thomas, G.; Simmen, T. The subcellular distribution of calnexin is mediated by pacs-2. Mol. Biol. Cell 2008, 19, 2777–2788. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, B.; Zheng, P.; Feng, H.; Chen, L.; Zhang, J.; Zhang, C.; Zhang, G.; Teng, J.; Chen, J. Silkworm coatomers and their role in tube expansion of posterior silkgland. PLoS ONE 2010, 5, e13252. [Google Scholar] [CrossRef]
- Petrosyan, A.; Cheng, P.W. A non-enzymatic function of golgi glycosyltransferases: Mediation of golgi fragmentation by interaction with non-muscle myosin iia. Glycobiology 2013, 23, 690–708. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, X.; Nix, D.B.; Katoh, T.; Aoki, K.; Tiemeyer, M.; Wang, Y. Regulation of protein glycosylation and sorting by the golgi matrix proteins grasp55/65. Nat. Commun. 2013, 4, 1659. [Google Scholar] [CrossRef]
- Puthenveedu, M.A.; Bachert, C.; Puri, S.; Lanni, F.; Linstedt, A.D. Gm130 and grasp65-dependent lateral cisternal fusion allows uniform golgi-enzyme distribution. Nat. Cell Biol. 2006, 8, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Mardones, G.A.; Snyder, C.M.; Howell, K.E. Cis-golgi matrix proteins move directly to endoplasmic reticulum exit sites by association with tubules. Mol. Biol. Cell 2006, 17, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Kellokumpu, S.; Sormunen, R.; Kellokumpu, I. Abnormal glycosylation and altered golgi structure in colorectal cancer: Dependence on intra-golgi ph. FEBS Lett. 2002, 516, 217–224. [Google Scholar] [CrossRef]
- Petrosyan, A.; Holzapfel, M.S.; Muirhead, D.E.; Cheng, P.W. Restoration of compact golgi morphology in advanced prostate cancer enhances susceptibility to galectin-1-induced apoptosis by modifying mucin o-glycan synthesis. Mol. Cancer Res. 2014, 12, 1704–1716. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Constructing a golgi complex. J. Cell Biol. 2001, 155, 873–875. [Google Scholar] [CrossRef]
- Ward, T.H.; Polishchuk, R.S.; Caplan, S.; Hirschberg, K.; Lippincott-Schwartz, J. Maintenance of golgi structure and function depends on the integrity of er export. J. Cell Biol. 2001, 155, 557–570. [Google Scholar] [CrossRef]
- Barr, F.A.; Short, B. Golgins in the structure and dynamics of the golgi apparatus. Curr. Opin. Cell Biol. 2003, 15, 405–413. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Hauri, H.P. The er-golgi intermediate compartment (ergic): In search of its identity and function. J. Cell Sci. 2006, 119, 2173–2183. [Google Scholar] [CrossRef]
- Sonnichsen, B.; Lowe, M.; Levine, T.; Jamsa, E.; Dirac-Svejstrup, B.; Warren, G. A role for giantin in docking copi vesicles to golgi membranes. J. Cell Biol. 1998, 140, 1013–1021. [Google Scholar] [CrossRef]
- Dirac-Svejstrup, A.B.; Shorter, J.; Waters, M.G.; Warren, G. Phosphorylation of the vesicle-tethering protein p115 by a casein kinase ii-like enzyme is required for golgi reassembly from isolated mitotic fragments. J. Cell Biol. 2000, 150, 475–488. [Google Scholar] [CrossRef]
- Lin, C.Y.; Madsen, M.L.; Yarm, F.R.; Jang, Y.J.; Liu, X.; Erikson, R.L. Peripheral golgi protein grasp65 is a target of mitotic polo-like kinase (plk) and cdc2. Proc. Natl. Acad. Sci. USA 2000, 97, 12589–12594. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.; Gonatas, N.K.; Warren, G. The mitotic phosphorylation cycle of the cis-golgi matrix protein gm130. J. Cell Biol. 2000, 149, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A.; Ali, M.F.; Cheng, P.W. Glycosyltransferase-specific golgi-targeting mechanisms. J. Biol. Chem. 2012, 287, 37621–37627. [Google Scholar] [CrossRef] [PubMed]
- Linstedt, A.D.; Hauri, H.P. Giantin, a novel conserved golgi membrane protein containing a cytoplasmic domain of at least 350 kda. Mol. Biol. Cell 1993, 4, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Linstedt, A.D.; Foguet, M.; Renz, M.; Seelig, H.P.; Glick, B.S.; Hauri, H.P. A c-terminally-anchored golgi protein is inserted into the endoplasmic reticulum and then transported to the golgi apparatus. Proc. Natl. Acad. Sci. USA 1995, 92, 5102–5105. [Google Scholar] [CrossRef]
- Lowe, M.; Lane, J.D.; Woodman, P.G.; Allan, V.J. Caspase-mediated cleavage of syntaxin 5 and giantin accompanies inhibition of secretory traffic during apoptosis. J. Cell Sci. 2004, 117, 1139–1150. [Google Scholar] [CrossRef]
- Nozawa, K.; Fritzler, M.J.; von Muhlen, C.A.; Chan, E.K. Giantin is the major golgi autoantigen in human anti-golgi complex sera. Arthritis Res. Ther. 2004, 6, R95–R102. [Google Scholar] [CrossRef]
- Nozawa, K.; Casiano, C.A.; Hamel, J.C.; Molinaro, C.; Fritzler, M.J.; Chan, E.K. Fragmentation of golgi complex and golgi autoantigens during apoptosis and necrosis. Arthritis Res. 2002, 4, R3. [Google Scholar] [CrossRef]
- Pelletier, L.; Jokitalo, E.; Warren, G. The effect of golgi depletion on exocytic transport. Nat. Cell Biol. 2000, 2, 840–846. [Google Scholar] [CrossRef]
- Koreishi, M.; Gniadek, T.J.; Yu, S.; Masuda, J.; Honjo, Y.; Satoh, A. The golgin tether giantin regulates the secretory pathway by controlling stack organization within golgi apparatus. PLoS ONE 2013, 8, e59821. [Google Scholar] [CrossRef]
- Casey, C.A.; Bhat, G.; Holzapfel, M.S.; Petrosyan, A. Study of ethanol-induced golgi disorganization reveals the potential mechanism of alcohol-impaired n-glycosylation. Alcohol. Clin. Exp. Res. 2016, 40, 2573–2590. [Google Scholar] [CrossRef] [PubMed]
- Manca, S.; Frisbie, C.P.; LaGrange, C.A.; Casey, C.A.; Riethoven, J.M.; Petrosyan, A. The role of alcohol-induced golgi fragmentation for androgen receptor signaling in prostate cancer. Mol. Cancer Res. 2019, 17, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Kubyshkin, A.V.; Fomochkina, I.I.; Petrosyan, A.M. The impact of alcohol on pro-metastatic n-glycosylation in prostate cancer. Krim. Z. Eksp. Klin. Med. 2018, 8, 11–20. [Google Scholar]
- Stevenson, N.L.; Bergen, D.J.M.; Skinner, R.E.H.; Kague, E.; Martin-Silverstone, E.; Robson Brown, K.A.; Hammond, C.L.; Stephens, D.J. Giantin-knockout models reveal a feedback loop between golgi function and glycosyltransferase expression. J. Cell Sci. 2017, 130, 4132–4143. [Google Scholar] [CrossRef] [PubMed]
- Casey, C.A.; Thomes, P.; Manca, S.; Petrosyan, A. Giantin is required for post-alcohol recovery of golgi in liver cells. Biomolecules 2018, 8, 150. [Google Scholar] [CrossRef]
- Dupre, D.J.; Robitaille, M.; Ethier, N.; Villeneuve, L.R.; Mamarbachi, A.M.; Hebert, T.E. Seven transmembrane receptor core signaling complexes are assembled prior to plasma membrane trafficking. J. Biol. Chem. 2006, 281, 34561–34573. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Kaufmann, R.; Piontek, J.; Grull, F.; Kirchgessner, M.; Rossa, J.; Wolburg, H.; Blasig, I.E.; Cremer, C. Visualization and quantitative analysis of reconstituted tight junctions using localization microscopy. PLoS ONE 2012, 7, e31128. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]
- Cembrowski, G.S.; Westgard, J.O.; Conover, W.J.; Toren, E.C., Jr. Statistical analysis of method comparison data. Testing normality. Am. J. Clin. Pathol. 1979, 72, 21–26. [Google Scholar] [CrossRef]
- Gibbons, J.D.; Chakraborti, S. Nonparametric Statistical Inference, 5th ed.; Taylor & Francis: Boca Raton, FL, USA, 2011; p. 630. [Google Scholar]
- Lee, S.A.; Kim, Y.J.; Lee, C.S. Brefeldin a induces apoptosis by activating the mitochondrial and death receptor pathways and inhibits focal adhesion kinase-mediated cell invasion. Basic Clin. Pharm. Toxicol. 2013, 113, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Tittle, T.V.; Allen, H.; Maziarz, R.T. Brefeldin a-mediated apoptosis requires the activation of caspases and is inhibited by bcl-2. Exp. Cell Res. 1998, 245, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Sohda, M.; Tashiro, A.; Sato, H.; Ikehara, Y. An essential cytoplasmic domain for the golgi localization of coiled-coil proteins with a cooh-terminal membrane anchor. J. Biol. Chem. 2001, 276, 6867–6873. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A.; Ali, M.F.; Verma, S.K.; Cheng, H.; Cheng, P.W. Non-muscle myosin iia transports a golgi glycosyltransferase to the endoplasmic reticulum by binding to its cytoplasmic tail. Int. J. Biochem. Cell Biol. 2012, 44, 1153–1165. [Google Scholar] [CrossRef]
- Sakata, N.; Phillips, T.E.; Dixon, J.L. Distribution, transport, and degradation of apolipoprotein b-100 in hepg2 cells. J. Lipid Res. 2001, 42, 1947–1958. [Google Scholar]
- Dusseljee, S.; Wubbolts, R.; Verwoerd, D.; Tulp, A.; Janssen, H.; Calafat, J.; Neefjes, J. Removal and degradation of the free mhc class ii beta chain in the endoplasmic reticulum requires proteasomes and is accelerated by bfa. J. Cell Sci. 1998, 111 Pt 15, 2217–2226. [Google Scholar]
- Puri, S.; Telfer, H.; Velliste, M.; Murphy, R.F.; Linstedt, A.D. Dispersal of golgi matrix proteins during mitotic golgi disassembly. J. Cell Sci. 2004, 117, 451–456. [Google Scholar] [CrossRef]
- Puthenveedu, M.A.; Linstedt, A.D. Evidence that golgi structure depends on a p115 activity that is independent of the vesicle tether components giantin and gm130. J. Cell Biol. 2001, 155, 227–238. [Google Scholar] [CrossRef]
- Hendricks, L.C.; McClanahan, S.L.; McCaffery, M.; Palade, G.E.; Farquhar, M.G. Golgi proteins persist in the tubulovesicular remnants found in brefeldin a-treated pancreatic acinar cells. Eur. J. Cell Biol. 1992, 58, 202–213. [Google Scholar]
- Seemann, J.; Jokitalo, E.; Pypaert, M.; Warren, G. Matrix proteins can generate the higher order architecture of the golgi apparatus. Nature 2000, 407, 1022–1026. [Google Scholar] [CrossRef]
- Soderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstrale, K.; Leuchowius, K.J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.; Bahram, F.; Li, X.; Gualandi, L.; Koch, S.; Jarvius, M.; Soderberg, O.; Anisimov, A.; Kholova, I.; Pytowski, B.; et al. Vegf receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J. 2010, 29, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Van Craenenbroeck, K.; Romero-Fernandez, W.; Guidolin, D.; Woods, A.S.; Rivera, A.; Haegeman, G.; Agnati, L.F.; Tarakanov, A.O.; Fuxe, K. Dopamine d2 and d4 receptor heteromerization and its allosteric receptor-receptor interactions. Biochem. Biophys. Res. Commun. 2011, 404, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Wang, R.A.; Mazumdar, A.; Talukder, A.H.; Mandal, M.; Yang, Z.; Bagheri-Yarmand, R.; Sahin, A.; Hortobagyi, G.; Adam, L.; et al. A naturally occurring mta1 variant sequesters oestrogen receptor-alpha in the cytoplasm. Nature 2002, 418, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Perez, M.E.; Dillingham, M.S.; Moreno-Herrero, F. Afm volumetric methods for the characterization of proteins and nucleic acids. Methods 2013, 60, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Lushnikov, A.Y.; Potaman, V.N.; Oussatcheva, E.A.; Sinden, R.R.; Lyubchenko, Y.L. DNA strand arrangement within the sfii-DNA complex: Atomic force microscopy analysis. Biochemistry 2006, 45, 152–158. [Google Scholar] [CrossRef][Green Version]
- Shlyakhtenko, L.S.; Lushnikov, A.Y.; Li, M.; Lackey, L.; Harris, R.S.; Lyubchenko, Y.L. Atomic force microscopy studies provide direct evidence for dimerization of the hiv restriction factor apobec3g. J. Biol. Chem. 2011, 286, 3387–3395. [Google Scholar] [CrossRef]
- Lyubchenko, Y.L.; Shlyakhtenko, L.S. Afm for analysis of structure and dynamics of DNA and protein-DNA complexes. Methods 2009, 47, 206–213. [Google Scholar] [CrossRef]
- Kasap, M.; Thomas, S.; Danaher, E.; Holton, V.; Jiang, S.; Storrie, B. Dynamic nucleation of golgi apparatus assembly from the endoplasmic reticulum in interphase hela cells. Traffic 2004, 5, 595–605. [Google Scholar] [CrossRef]
- Jiang, S.; Rhee, S.W.; Gleeson, P.A.; Storrie, B. Capacity of the golgi apparatus for cargo transport prior to complete assembly. Mol. Biol. Cell 2006, 17, 4105–4117. [Google Scholar] [CrossRef]
- Szul, T.; Sztul, E. Copii and copi traffic at the er-golgi interface. Physiology 2011, 26, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Menarguez, J.A.; Prekeris, R.; Oorschot, V.M.; Scheller, R.; Slot, J.W.; Geuze, H.J.; Klumperman, J. Peri-golgi vesicles contain retrograde but not anterograde proteins consistent with the cisternal progression model of intra-golgi transport. J. Cell Biol. 2001, 155, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Gosavi, P.; Houghton, F.J.; McMillan, P.J.; Hanssen, E.; Gleeson, P.A. The golgi ribbon in mammalian cells negatively regulates autophagy by modulating mtor activity. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Taneja, T.K.; Ma, D.; Kim, B.Y.; Welling, P.A. Golgin-97 targets ectopically expressed inward rectifying potassium channel, kir2.1, to the trans-golgi network in cos-7 cells. Front. Physiol. 2018, 9, 1070. [Google Scholar] [CrossRef]
- Lieu, Z.Z.; Lock, J.G.; Hammond, L.A.; La Gruta, N.L.; Stow, J.L.; Gleeson, P.A. A trans-golgi network golgin is required for the regulated secretion of tnf in activated macrophages in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 3351–3356. [Google Scholar] [CrossRef]
- Reddy, J.V.; Burguete, A.S.; Sridevi, K.; Ganley, I.G.; Nottingham, R.M.; Pfeffer, S.R. A functional role for the gcc185 golgin in mannose 6-phosphate receptor recycling. Mol. Biol. Cell 2006, 17, 4353–4363. [Google Scholar] [CrossRef]
- Yamane, J.; Kubo, A.; Nakayama, K.; Yuba-Kubo, A.; Katsuno, T.; Tsukita, S.; Tsukita, S. Functional involvement of tmf/ara160 in rab6-dependent retrograde membrane traffic. Exp. Cell Res. 2007, 313, 3472–3485. [Google Scholar] [CrossRef]
- Sogaard, M.; Tani, K.; Ye, R.R.; Geromanos, S.; Tempst, P.; Kirchhausen, T.; Rothman, J.E.; Sollner, T. A rab protein is required for the assembly of snare complexes in the docking of transport vesicles. Cell 1994, 78, 937–948. [Google Scholar] [CrossRef]
- Liu, S.; Storrie, B. Are rab proteins the link between golgi organization and membrane trafficking? Cell Mol. Life Sci. 2012, 69, 4093–4106. [Google Scholar] [CrossRef]
- Bard, F.; Casano, L.; Mallabiabarrena, A.; Wallace, E.; Saito, K.; Kitayama, H.; Guizzunti, G.; Hu, Y.; Wendler, F.; Dasgupta, R.; et al. Functional genomics reveals genes involved in protein secretion and golgi organization. Nature 2006, 439, 604–607. [Google Scholar] [CrossRef]
- Dejgaard, S.Y.; Murshid, A.; Erman, A.; Kizilay, O.; Verbich, D.; Lodge, R.; Dejgaard, K.; Ly-Hartig, T.B.; Pepperkok, R.; Simpson, J.C.; et al. Rab18 and rab43 have key roles in er-golgi trafficking. J. Cell Sci. 2008, 121, 2768–2781. [Google Scholar] [CrossRef] [PubMed]
- Miserey-Lenkei, S.; Chalancon, G.; Bardin, S.; Formstecher, E.; Goud, B.; Echard, A. Rab and actomyosin-dependent fission of transport vesicles at the golgi complex. Nat. Cell Biol. 2010, 12, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Satpute-Krishnan, P.; Seo, A.Y.; Burnette, D.T.; Patterson, G.H.; Lippincott-Schwartz, J. Er trapping reveals golgi enzymes continually revisit the er through a recycling pathway that controls golgi organization. Proc. Natl. Acad. Sci. USA 2015, 112, E6752–E6761. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A.; Casey, C.A.; Cheng, P.W. The role of rab6a and phosphorylation of non-muscle myosin iia tailpiece in alcohol-induced golgi disorganization. Sci. Rep. 2016, 6, 31962. [Google Scholar] [CrossRef]
- Rosing, M.; Ossendorf, E.; Rak, A.; Barnekow, A. Giantin interacts with both the small gtpase rab6 and rab1. Exp. Cell Res. 2007, 313, 2318–2325. [Google Scholar] [CrossRef]
- Cluett, E.B.; de Figueiredo, P.; Bechler, M.E.; Thorsen, K.D.; Brown, W.J. Reconstitution of phospholipase a2-dependent golgi membrane tubules. Methods Mol. Biol. 2016, 1496, 75–90. [Google Scholar]
- Matsuto, M.; Kano, F.; Murata, M. Reconstitution of the targeting of rab6a to the golgi apparatus in semi-intact hela cells: A role of bicd2 in stabilizing rab6a on golgi membranes and a concerted role of rab6a/bicd2 interactions in golgi-to-er retrograde transport. Biochim. Biophys. Acta 2015, 1853, 2592–2609. [Google Scholar] [CrossRef]
- Del Nery, E.; Miserey-Lenkei, S.; Falguieres, T.; Nizak, C.; Johannes, L.; Perez, F.; Goud, B. Rab6a and rab6a’ gtpases play non-overlapping roles in membrane trafficking. Traffic 2006, 7, 394–407. [Google Scholar] [CrossRef]
- Micaroni, M.; Stanley, A.C.; Khromykh, T.; Venturato, J.; Wong, C.X.; Lim, J.P.; Marsh, B.J.; Storrie, B.; Gleeson, P.A.; Stow, J.L. Rab6a/a’ are important golgi regulators of pro-inflammatory tnf secretion in macrophages. PLoS ONE 2013, 8, e57034. [Google Scholar] [CrossRef]
- Young, J.; Stauber, T.; del Nery, E.; Vernos, I.; Pepperkok, R.; Nilsson, T. Regulation of microtubule-dependent recycling at the trans-golgi network by rab6a and rab6a. Mol. Biol. Cell 2005, 16, 162–177. [Google Scholar] [CrossRef]
- Nakamura, N.; Lowe, M.; Levine, T.P.; Rabouille, C.; Warren, G. The vesicle docking protein p115 binds gm130, a cis-golgi matrix protein, in a mitotically regulated manner. Cell 1997, 89, 445–455. [Google Scholar] [CrossRef]
- Barr, F.A.; Puype, M.; Vandekerckhove, J.; Warren, G. Grasp65, a protein involved in the stacking of golgi cisternae. Cell 1997, 91, 253–262. [Google Scholar] [CrossRef]
- Lesa, G.M.; Seemann, J.; Shorter, J.; Vandekerckhove, J.; Warren, G. The amino-terminal domain of the golgi protein giantin interacts directly with the vesicle-tethering protein p115. J. Biol. Chem. 2000, 275, 2831–2836. [Google Scholar] [CrossRef] [PubMed]
- Asante, D.; Maccarthy-Morrogh, L.; Townley, A.K.; Weiss, M.A.; Katayama, K.; Palmer, K.J.; Suzuki, H.; Westlake, C.J.; Stephens, D.J. A role for the golgi matrix protein giantin in ciliogenesis through control of the localization of dynein-2. J. Cell Sci. 2013, 126, 5189–5197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Seemann, J.; Pypaert, M.; Shorter, J.; Warren, G. A direct role for grasp65 as a mitotically regulated golgi stacking factor. EMBO J. 2003, 22, 3279–3290. [Google Scholar] [CrossRef]
- Wang, Y.; Satoh, A.; Warren, G. Mapping the functional domains of the golgi stacking factor grasp65. J. Biol. Chem. 2005, 280, 4921–4928. [Google Scholar] [CrossRef]
- Feng, Y.; Yu, W.; Li, X.; Lin, S.; Zhou, Y.; Hu, J.; Liu, X. Structural insight into golgi membrane stacking by grasp65 and grasp55 proteins. J. Biol. Chem. 2013, 288, 28418–28427. [Google Scholar]
- Xiang, Y.; Wang, Y. Grasp55 and grasp65 play complementary and essential roles in golgi cisternal stacking. J. Cell Biol. 2010, 188, 237–251. [Google Scholar] [CrossRef]
- Yoshimura, S.; Yamamoto, A.; Misumi, Y.; Sohda, M.; Barr, F.A.; Fujii, G.; Shakoori, A.; Ohno, H.; Mihara, K.; Nakamura, N. Dynamics of golgi matrix proteins after the blockage of er to golgi transport. J. Biochem. 2004, 135, 201–216. [Google Scholar] [CrossRef]
- Kim, J.; Noh, S.H.; Piao, H.; Kim, D.H.; Kim, K.; Cha, J.S.; Chung, W.Y.; Cho, H.S.; Kim, J.Y.; Lee, M.G. Monomerization and er relocalization of grasp is a requisite for unconventional secretion of cftr. Traffic 2016, 17, 733–753. [Google Scholar] [CrossRef]
- Ignashkova, T.I.; Gendarme, M.; Peschk, K.; Eggenweiler, H.M.; Lindemann, R.K.; Reiling, J.H. Cell survival and protein secretion associated with golgi integrity in response to golgi stress-inducing agents. Traffic 2017, 18, 530–544. [Google Scholar] [PubMed]
- Trucco, A.; Polishchuk, R.S.; Martella, O.; Di Pentima, A.; Fusella, A.; Di Giandomenico, D.; San Pietro, E.; Beznoussenko, G.V.; Polishchuk, E.V.; Baldassarre, M.; et al. Secretory traffic triggers the formation of tubular continuities across golgi sub-compartments. Nat. Cell Biol. 2004, 6, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Rambourg, A.; Clermont, Y.; Kepes, F. Modulation of the golgi apparatus in saccharomyces cerevisiae sec7 mutants as seen by three-dimensional electron microscopy. Anat. Rec. 1993, 237, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Mironov, A.A.; Sesorova, I.S.; Seliverstova, E.V.; Beznoussenko, G.V. Different golgi ultrastructure across species and tissues: Implications under functional and pathological conditions, and an attempt at classification. Tissue Cell 2017, 49, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Lauzon, A.M.; Fagnant, P.M.; Warshaw, D.M.; Trybus, K.M. Coiled-coil unwinding at the smooth muscle myosin head-rod junction is required for optimal mechanical performance. Biophys. J. 2001, 80, 1900–1904. [Google Scholar]
- Cheung, P.Y.; Limouse, C.; Mabuchi, H.; Pfeffer, S.R. Protein flexibility is required for vesicle tethering at the golgi. Elife 2015, 4, e12790. [Google Scholar]
- Knight, P.J.; Thirumurugan, K.; Xu, Y.; Wang, F.; Kalverda, A.P.; Stafford, W.F., 3rd; Sellers, J.R.; Peckham, M. The predicted coiled-coil domain of myosin 10 forms a novel elongated domain that lengthens the head. J. Biol. Chem. 2005, 280, 34702–34708. [Google Scholar]
- Nizak, C.; Martin-Lluesma, S.; Moutel, S.; Roux, A.; Kreis, T.E.; Goud, B.; Perez, F. Recombinant antibodies against subcellular fractions used to track endogenous golgi protein dynamics in vivo. Traffic 2003, 4, 739–753. [Google Scholar] [CrossRef]
- Satoh, A.; Hayashi-Nishino, M.; Shakuno, T.; Masuda, J.; Koreishi, M.; Murakami, R.; Nakamura, Y.; Nakamura, T.; Abe-Kanoh, N.; Honjo, Y.; et al. The golgin protein giantin regulates interconnections between golgi stacks. Front. Cell Dev. Biol. 2019, 7, 160. [Google Scholar] [CrossRef]
- Lee, I.; Tiwari, N.; Dunlop, M.H.; Graham, M.; Liu, X.; Rothman, J.E. Membrane adhesion dictates golgi stacking and cisternal morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 1849–1854. [Google Scholar] [CrossRef]
- Bergbrede, T.; Pylypenko, O.; Rak, A.; Alexandrov, K. Structure of the extremely slow gtpase rab6a in the gtp bound form at 1.8a resolution. J. Struct. Biol. 2005, 152, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.M.; Valderrama, F.; Castel, S.; Magdalena, J.; Tomas, M.; Hosoya, H.; Renau-Piqueras, J.; Malhotra, V.; Egea, G. Myosin motors and not actin comets are mediators of the actin-based golgi-to-endoplasmic reticulum protein transport. Mol. Biol. Cell 2003, 14, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.H.; Seemann, J. Unraveling the golgi ribbon. Traffic 2010, 11, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Hong, S.H.; Jiang, H.L.; Minai-Tehrani, A.; Yu, K.N.; Lee, J.H.; Kim, J.E.; Shin, J.Y.; Kang, B.; Park, S.; et al. Golga2/gm130, cis-golgi matrix protein, is a novel target of anticancer gene therapy. Mol. 2012, 20, 2052–2063. [Google Scholar] [CrossRef]
- Quintero, C.A.; Valdez-Taubas, J.; Ferrari, M.L.; Haedo, S.D.; Maccioni, H.J. Calsenilin and calp interact with the cytoplasmic tail of udp-gal:Ga2/gm2/gd2 beta-1,3-galactosyltransferase. Biochem. J. 2008, 412, 19–26. [Google Scholar] [CrossRef]
- Pokrovskaya, I.D.; Willett, R.; Smith, R.D.; Morelle, W.; Kudlyk, T.; Lupashin, V.V. Conserved oligomeric golgi complex specifically regulates the maintenance of golgi glycosylation machinery. Glycobiology 2011, 21, 1554–1569. [Google Scholar] [CrossRef]
- Gill, D.J.; Chia, J.; Senewiratne, J.; Bard, F. Regulation of o-glycosylation through golgi-to-er relocation of initiation enzymes. J. Cell Biol. 2010, 189, 843–858. [Google Scholar] [CrossRef]
- Nilsson, T.; Rabouille, C.; Hui, N.; Watson, R.; Warren, G. The role of the membrane-spanning domain and stalk region of n-acetylglucosaminyltransferase i in retention, kin recognition and structural maintenance of the golgi apparatus in hela cells. J. Cell Sci. 1996, 109 Pt 7, 1975–1989. [Google Scholar]
- Dong, Z.; Zuber, C.; Pierce, M.; Stanley, P.; Roth, J. Reduction in golgi apparatus dimension in the absence of a residential protein, n-acetylglucosaminyltransferase v. Histochem. Cell Biol. 2014, 141, 153–164. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Fukuda, M.N. Golgi retention mechanism of beta-1,4-galactosyltransferase. Membrane-spanning domain-dependent homodimerization and association with alpha- and beta-tubulins. J. Biol. Chem. 1995, 270, 12170–12176. [Google Scholar] [CrossRef]
- Wassler, M.J.; Foote, C.I.; Gelman, I.H.; Shur, B.D. Functional interaction between the ssecks scaffolding protein and the cytoplasmic domain of beta1,4-galactosyltransferase. J. Cell Sci. 2001, 114, 2291–2300. [Google Scholar] [PubMed]
- Petrosyan, A.; Ali, M.F.; Cheng, P.W. Keratin 1 plays a critical role in golgi localization of core 2 n-acetylglucosaminyltransferase m via interaction with its cytoplasmic tail. J. Biol. Chem. 2015, 290, 6256–6269. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.F.; Chachadi, V.B.; Petrosyan, A.; Cheng, P.W. Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling golgi localization of core 2 n-acetylglucosaminyltransferase 1. J. Biol. Chem. 2012, 287, 39564–39577. [Google Scholar] [CrossRef] [PubMed]
- Marra, P.; Salvatore, L.; Mironov, A., Jr.; Di Campli, A.; Di Tullio, G.; Trucco, A.; Beznoussenko, G.; Mironov, A.; De Matteis, M.A. The biogenesis of the golgi ribbon: The roles of membrane input from the er and of gm130. Mol. Biol. Cell 2007, 18, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.R.; Chay, C.H.; Pienta, K.J. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia 2002, 4, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Li, J.; Lu, H.; Akech, J.; Pratap, J.; Wang, T.; Zerlanko, B.J.; FitzGerald, T.J.; Jiang, Z.; Birbe, R.; et al. Integrin alphavbeta6 promotes an osteolytic program in cancer cells by upregulating mmp2. Cancer Res. 2014, 74, 1598–1608. [Google Scholar] [CrossRef]
- List, K.; Szabo, R.; Molinolo, A.; Sriuranpong, V.; Redeye, V.; Murdock, T.; Burke, B.; Nielsen, B.S.; Gutkind, J.S.; Bugge, T.H. Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev. 2005, 19, 1934–1950. [Google Scholar] [CrossRef]
- Uhland, K. Matriptase and its putative role in cancer. Cell Mol. Life Sci. 2006, 63, 2968–2978. [Google Scholar] [CrossRef]
- Bianconi, D.; Unseld, M.; Prager, G.W. Integrins in the spotlight of cancer. Int. J. Mol. Sci. 2016, 17, 2037. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Machamer, C.E. The golgi complex in stress and death. Front. Neurosci. 2015, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Lujan, H.D.; Marotta, A.; Mowatt, M.R.; Sciaky, N.; Lippincott-Schwartz, J.; Nash, T.E. Developmental induction of golgi structure and function in the primitive eukaryote giardia lamblia. J. Biol. Chem. 1995, 270, 4612–4618. [Google Scholar] [CrossRef] [PubMed]
- May, J.A.; Ratan, H.; Glenn, J.R.; Losche, W.; Spangenberg, P.; Heptinstall, S. Gpiib-iiia antagonists cause rapid disaggregation of platelets pre-treated with cytochalasin d. Evidence that the stability of platelet aggregates depends on normal cytoskeletal assembly. Platelets 1998, 9, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R.; Tartakoff, A.M. The response of the golgi complex to microtubule alterations: The roles of metabolic energy and membrane traffic in golgi complex organization. J. Cell Biol. 1989, 109, 2081–2088. [Google Scholar] [CrossRef]
- Irurzun, A.; Perez, L.; Carrasco, L. Brefeldin a blocks protein glycosylation and rna replication of vesicular stomatitis virus. FEBS Lett. 1993, 336, 496–500. [Google Scholar] [CrossRef]
- Liu, L.; Bastien, N.; Li, Y. Intracellular processing, glycosylation, and cell surface expression of human metapneumovirus attachment glycoprotein. J. Virol. 2007, 81, 13435–13443. [Google Scholar] [CrossRef]
- Stallcup, K.C.; Raine, C.S.; Fields, B.N. Cytochalasin b inhibits the maturation of measles virus. Virology 1983, 124, 59–74. [Google Scholar] [CrossRef]
- Rios, R.M.; Bornens, M. The golgi apparatus at the cell centre. Curr. Opin. Cell Biol. 2003, 15, 60–66. [Google Scholar] [CrossRef]
- Linstedt, A.D. Positioning the golgi apparatus. Cell 2004, 118, 271–272. [Google Scholar] [CrossRef][Green Version]
- Cutrona, M.B.; Beznoussenko, G.V.; Fusella, A.; Martella, O.; Moral, P.; Mironov, A.A. Silencing of mammalian sar1 isoforms reveals copii-independent protein sorting and transport. Traffic 2013, 14, 691–708. [Google Scholar] [CrossRef]
- Matanis, T.; Akhmanova, A.; Wulf, P.; Del Nery, E.; Weide, T.; Stepanova, T.; Galjart, N.; Grosveld, F.; Goud, B.; De Zeeuw, C.I.; et al. Bicaudal-d regulates copi-independent golgi-er transport by recruiting the dynein-dynactin motor complex. Nat. Cell Biol. 2002, 4, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.C.; Nilsson, T.; Pepperkok, R. Biogenesis of tubular er-to-golgi transport intermediates. Mol. Biol. Cell 2006, 17, 723–737. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frisbie, C.P.; Lushnikov, A.Y.; Krasnoslobodtsev, A.V.; Riethoven, J.-J.M.; Clarke, J.L.; Stepchenkova, E.I.; Petrosyan, A. Post-ER Stress Biogenesis of Golgi Is Governed by Giantin. Cells 2019, 8, 1631. https://doi.org/10.3390/cells8121631
Frisbie CP, Lushnikov AY, Krasnoslobodtsev AV, Riethoven J-JM, Clarke JL, Stepchenkova EI, Petrosyan A. Post-ER Stress Biogenesis of Golgi Is Governed by Giantin. Cells. 2019; 8(12):1631. https://doi.org/10.3390/cells8121631
Chicago/Turabian StyleFrisbie, Cole P., Alexander Y. Lushnikov, Alexey V. Krasnoslobodtsev, Jean-Jack M. Riethoven, Jennifer L. Clarke, Elena I. Stepchenkova, and Armen Petrosyan. 2019. "Post-ER Stress Biogenesis of Golgi Is Governed by Giantin" Cells 8, no. 12: 1631. https://doi.org/10.3390/cells8121631
APA StyleFrisbie, C. P., Lushnikov, A. Y., Krasnoslobodtsev, A. V., Riethoven, J.-J. M., Clarke, J. L., Stepchenkova, E. I., & Petrosyan, A. (2019). Post-ER Stress Biogenesis of Golgi Is Governed by Giantin. Cells, 8(12), 1631. https://doi.org/10.3390/cells8121631