Identification of TRIM25 as a Negative Regulator of Caspase-2 Expression Reveals a Novel Target for Sensitizing Colon Carcinoma Cells to Intrinsic Apoptosis

 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. RNA Interference
2.4. Transient Overexpression of TRIM25
2.5. Western Blot Analysis
2.6. Mitochondrial Cytochrome C Release
2.7. qRT-PCR-Analysis
2.8. Ribonucleoprotein- (RNP) IP RT-PCR Assay
2.9. RNA Affinity Chromatography
2.10. Liquid Chromatography/Mass Spectrometry (LC/MS)
2.11. Separation of Polysomes from Translational Inactive RNP Granules
2.12. Confocal Microscopy
2.13. Statistical Analysis
3. Results
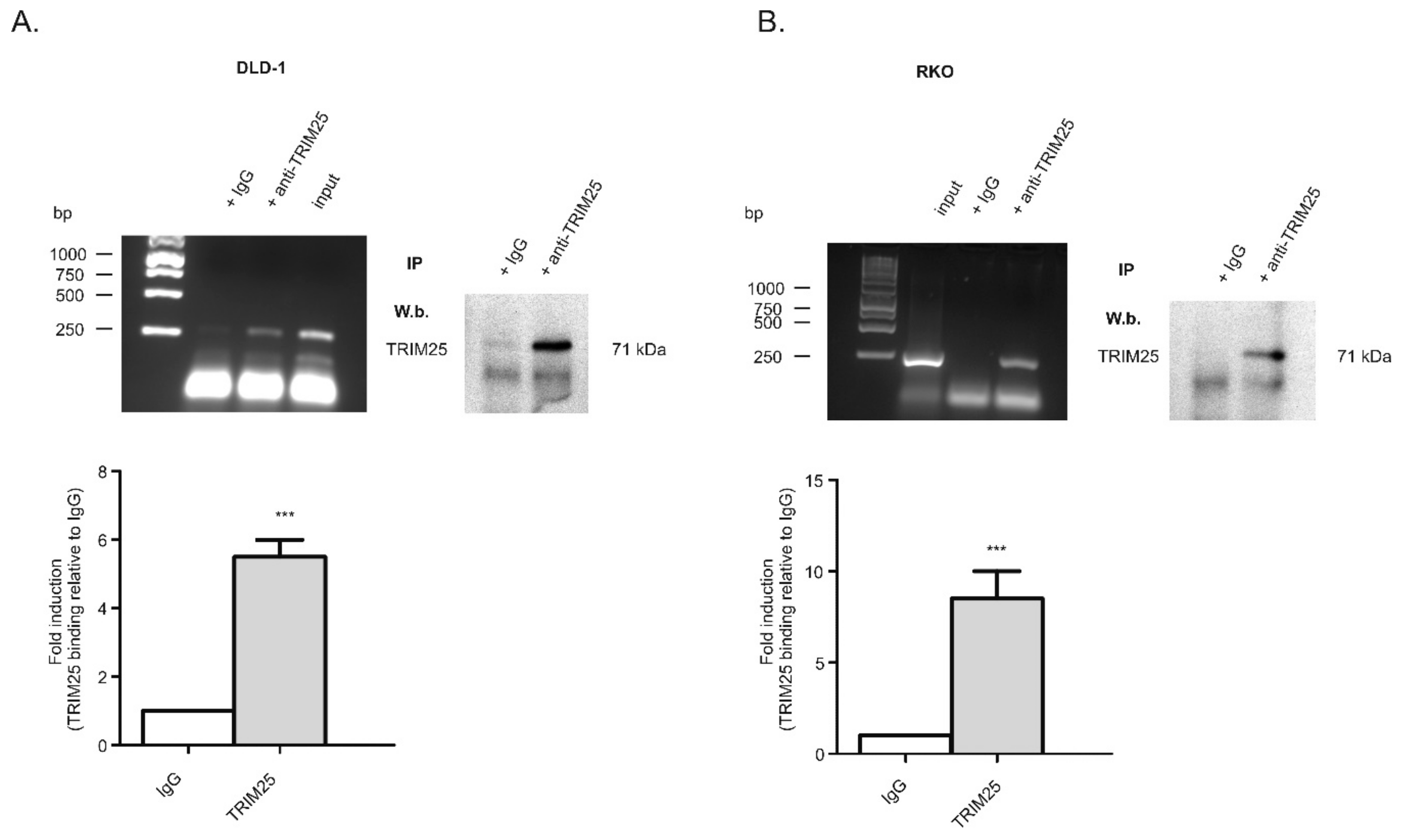
3.1. Identification of TRIM25 as a Novel Caspase-2 mRNA-Binding Protein
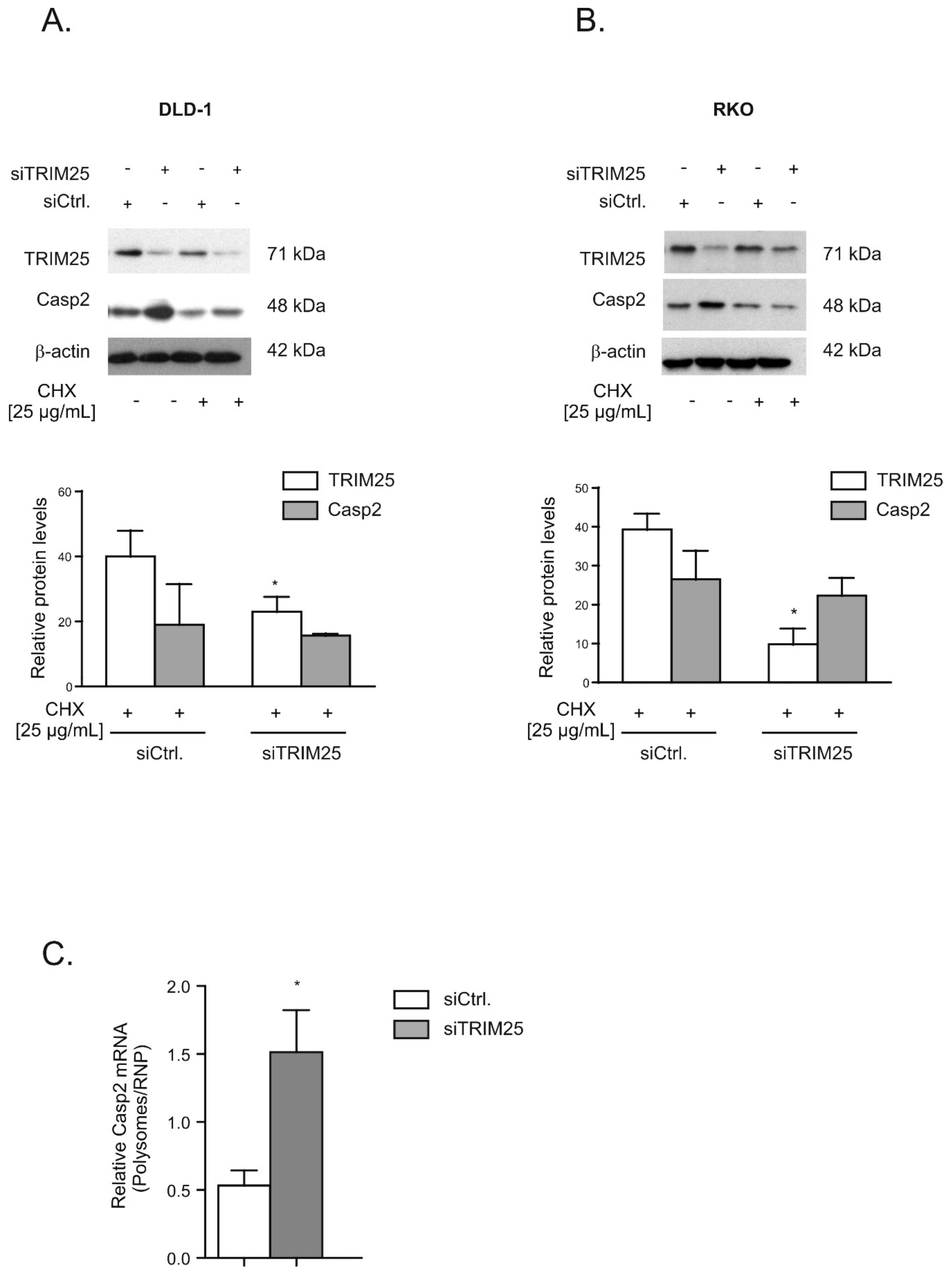
3.2. TRIM25 Negatively Interferes with Caspase-2 Translation
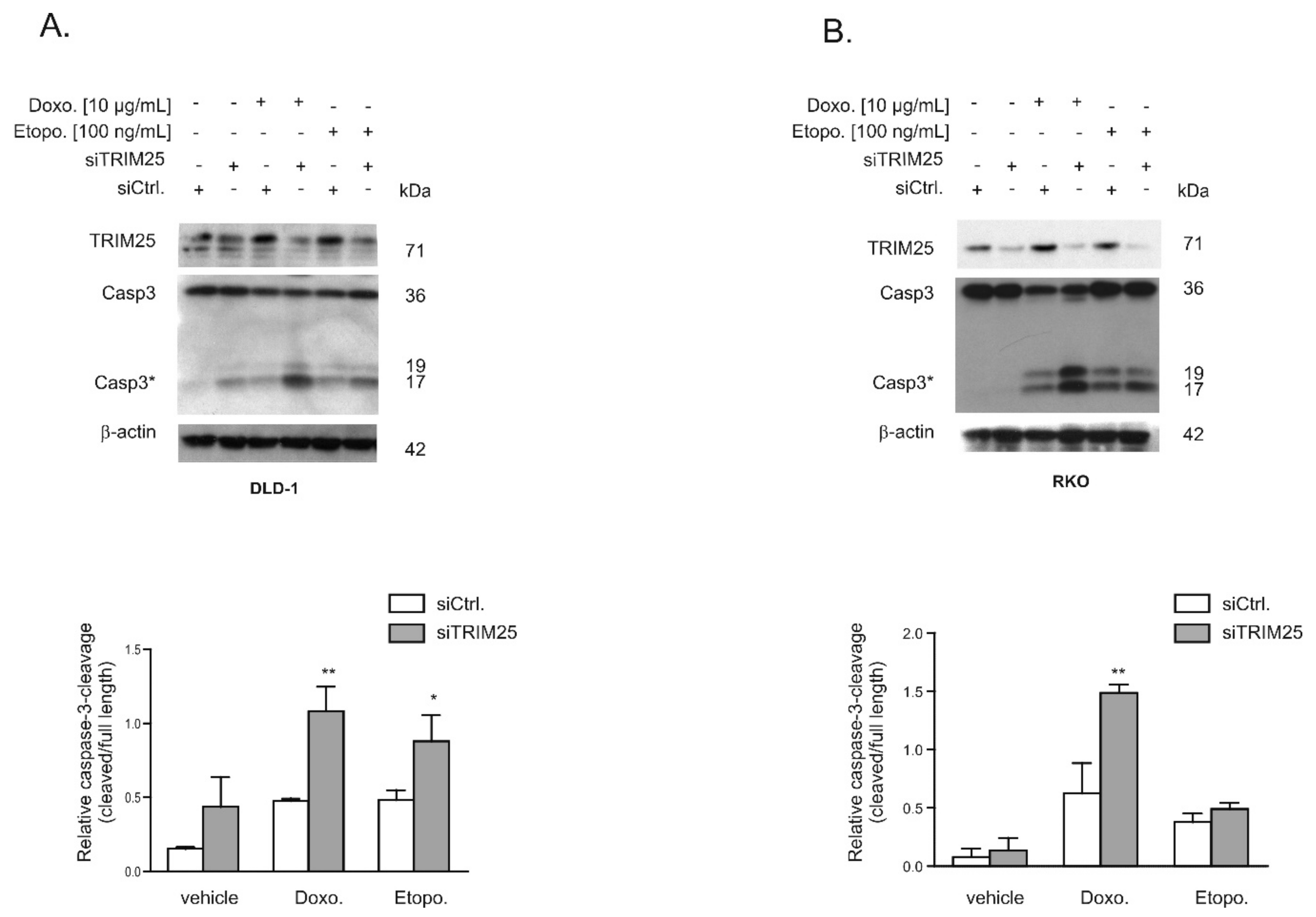
3.3. Knockdown of TRIM25 Sensitizes Colon Carcinoma Cells to Drug-Induced Apoptosis
3.4. Caspase-2 is Critical for Sensitization by TRIM25 Silencing
3.5. Increased TRIM25 Binding to 5′UTR of Caspase-2 by Doxorubicin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Flanagan, L.; Kehoe, J.; Fay, J.; Bacon, O.; Lindner, A.U.; Kay, E.W.; Deasy, J.; McNamara, D.A.; Prehn, J.H. High levels of X-linked Inhibitor-of-Apoptosis Protein (XIAP) are indicative of radio chemotherapy resistance in rectal cancer. Radiat. Oncol. 2015, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, T.; Rodel, F.; Beissbarth, T.; Conradi, L.C.; Rothe, H.; Homayounfar, K.; Wolff, H.A.; Ghadimi, B.M.; Yildirim, M.; Becker, H.; et al. Failure of downregulation of survivin following neoadjuvant radiochemotherapy in rectal cancer is associated with distant metastases and shortened survival. Clin Cancer Res. 2011, 17, 1623–1631. [Google Scholar] [CrossRef]
- McKenzie, S.; Kyprianou, N. Apoptosis evasion: The role of survival pathways in prostate cancer progression and therapeutic resistance. J. Cell Biochem. 2006, 97, 18–32. [Google Scholar] [CrossRef]
- Winkler, C.; Doller, A.; Imre, G.; Badawi, A.; Schmid, T.; Schulz, S.; Steinmeyer, N.; Pfeilschifter, J.; Rajalingam, K.; Eberhardt, W. Attenuation of the ELAV1-like protein HuR sensitizes adenocarcinoma cells to the intrinsic apoptotic pathway by increasing the translation of caspase-2L. Cell Death Dis. 2014, 5, e1321. [Google Scholar] [CrossRef]
- Badawi, A.; Hehlgans, S.; Pfeilschifter, J.; Rodel, F.; Eberhardt, W. Silencing of the mRNA-binding protein HuR increases the sensitivity of colorectal cancer cells to ionizing radiation through upregulation of caspase-2. Cancer Lett. 2017, 393, 103–112. [Google Scholar] [CrossRef]
- Badawi, A.; Biyanee, A.; Nasrullah, U.; Winslow, S.; Schmid, T.; Pfeilschifter, J.; Eberhardt, W. Inhibition of IRES-dependent translation of caspase-2 by HuR confers chemotherapeutic drug resistance in colon carcinoma cells. Oncotarget 2018, 9, 18367–18385. [Google Scholar] [CrossRef]
- Eberhardt, W.; Nasrullah, U.; Haeussler, K. Inhibition of Caspase-2 Translation by the mRNA Binding Protein HuR: A Novel Path of Therapy Resistance in Colon Carcinoma Cells? Cells 2019, 8, 797. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Sohm, B.; Bock, F.; Manzl, C.; Villunger, A. The enigma of caspase-2: The laymen′s view. Cell Death Differ. 2009, 16, 195–207. [Google Scholar] [CrossRef]
- Bouchier-Hayes, L. The role of caspase-2 in stress-induced apoptosis. J. Cell Mol. Med. 2010, 14, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Zhivotovsky, B. The unpredictable caspase-2: What can it do? Trends Cell Biol 2010, 20, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.H.; Taylor, R.; Dorstyn, L.; Cakouros, D.; Bouillet, P.; Kumar, S. A tumor suppressor function for caspase-2. Proc. Natl. Acad. Sci. USA 2009, 106, 5336–5341. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Lu, J.; Porollo, A.; Du, C. Tumor-suppressing function of caspase-2 requires catalytic site Cys-320 and site Ser-139 in mice. J. Biol. Chem. 2012, 287, 14792–14802. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; McCormick, L.; Janke, L.; Howard, A.; Bouchier-Hayes, L.; Green, D.R. Genetic deletion of caspase-2 accelerates MMTV/c-neu-driven mammary carcinogenesis in mice. Cell Death Differ. 2013, 20, 1174–1182. [Google Scholar] [CrossRef]
- Manzl, C.; Peintner, L.; Krumschnabel, G.; Bock, F.; Labi, V.; Drach, M.; Newbold, A.; Johnstone, R.; Villunger, A. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ. 2012, 19, 1722–1732. [Google Scholar] [CrossRef]
- Puccini, J.; Dorstyn, L.; Kumar, S. Caspase-2 as a tumour suppressor. Cell Death Differ. 2013, 20, 1133–1139. [Google Scholar] [CrossRef]
- Dawar, S.; Lim, Y.; Puccini, J.; White, M.; Thomas, P.; Bouchier-Hayes, L.; Green, D.R.; Dorstyn, L.; Kumar, S. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 2017, 36, 2704–2714. [Google Scholar] [CrossRef]
- Dorstyn, L.; Puccini, J.; Wilson, C.H.; Shalini, S.; Nicola, M.; Moore, S.; Kumar, S. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 2012, 19, 1288–1298. [Google Scholar] [CrossRef]
- Shalini, S.; Puccini, J.; Wilson, C.H.; Finnie, J.; Dorstyn, L.; Kumar, S. Caspase-2 protects against oxidative stress in vivo. Oncogene 2015, 34, 4995–5002. [Google Scholar] [CrossRef]
- Lopez-Garcia, C.; Sansregret, L.; Domingo, E.; McGranahan, N.; Hobor, S.; Birkbak, N.J.; Horswell, S.; Gronroos, E.; Favero, F.; Rowan, A.J.; et al. BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer. Cancer Cell 2017, 31, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Lee, J.W.; Kim, Y.J.; Soung, Y.H.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Lee, J.Y.; Lee, S.H. Loss of caspase-2, -6 and -7 expression in gastric cancers. APMIS 2004, 112, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Holleman, A.; den Boer, M.L.; Kazemier, K.M.; Beverloo, H.B.; von Bergh, A.R.; Janka-Schaub, G.E.; Pieters, R. Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood 2005, 106, 1817–1823. [Google Scholar] [CrossRef] [PubMed]
- Zohrabian, V.M.; Nandu, H.; Gulati, N.; Khitrov, G.; Zhao, C.; Mohan, A.; Demattia, J.; Braun, A.; Das, K.; Murali, R.; et al. Gene expression profiling of metastatic brain cancer. Oncol Rep. 2007, 18, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Heikel, G.; Choudhury, N.R.; Michlewski, G. The role of Trim25 in development, disease and RNA metabolism. Biochem. Soc. Trans. 2016, 44, 1045–1050. [Google Scholar] [CrossRef]
- Walsh, L.A.; Alvarez, M.J.; Sabio, E.Y.; Reyngold, M.; Makarov, V.; Mukherjee, S.; Lee, K.W.; Desrichard, A.; Turcan, S.; Dalin, M.G.; et al. An Integrated Systems Biology Approach Identifies TRIM25 as a Key Determinant of Breast Cancer Metastasis. Cell Rep. 2017, 20, 1623–1640. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. [Google Scholar] [CrossRef]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef]
- Doller, A.; Huwiler, A.; Muller, R.; Radeke, H.H.; Pfeilschifter, J.; Eberhardt, W. Protein kinase C alpha-dependent phosphorylation of the mRNA-stabilizing factor HuR: Implications for posttranscriptional regulation of cyclooxygenase-2. Mol. Biol. Cell 2007, 18, 2137–2148. [Google Scholar] [CrossRef]
- Doller, A.; Schulz, S.; Pfeilschifter, J.; Eberhardt, W. RNA-dependent association with myosin IIA promotes F-actin-guided trafficking of the ELAV-like protein HuR to polysomes. Nucleic Acids Res. 2013, 41, 9152–9167. [Google Scholar] [CrossRef]
- Schulz, S.; Doller, A.; Pendini, N.R.; Wilce, J.A.; Pfeilschifter, J.; Eberhardt, W. Domain-specific phosphomimetic mutation allows dissection of different protein kinase C (PKC) isotype-triggered activities of the RNA binding protein HuR. Cell Signal. 2013, 25, 2485–2495. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, Y.S.; Kim, D.K. Doxorubicin exerts cytotoxic effects through cell cycle arrest and Fas-mediated cell death. Pharmacology 2009, 84, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.C.; Yi, H.; Eichelbaum, K.; Fohr, S.; Fischer, B.; You, K.T.; Castello, A.; Krijgsveld, J.; Hentze, M.W.; Kim, V.N. The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1122–1130. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Humphries, C.G.; Ma, S.H.; Hutchinson, R.; Li, R.; Siddiqui, J.; Tomlins, S.A.; Raj, G.V.; Kittler, R. The ubiquitin ligase TRIM25 targets ERG for degradation in prostate cancer. Oncotarget 2016, 7, 64921–64931. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Heikel, G.; Trubitsyna, M.; Kubik, P.; Nowak, J.S.; Webb, S.; Granneman, S.; Spanos, C.; Rappsilber, J.; Castello, A.; et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. Bmc. Biol. 2017, 15, 105. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Nowak, J.S.; Zuo, J.; Rappsilber, J.; Spoel, S.H.; Michlewski, G. Trim25 Is an RNA-Specific Activator of Lin28a/TuT4-Mediated Uridylation. Cell Rep. 2014, 9, 1265–1272. [Google Scholar] [CrossRef]
- Kumar, S. Caspase 2 in apoptosis, the DNA damage response and tumour suppression: Enigma no more? Nat. Rev. Cancer 2009, 9, 897–903. [Google Scholar] [CrossRef]
- Zhang, P.; Elabd, S.; Hammer, S.; Solozobova, V.; Yan, H.; Bartel, F.; Inoue, S.; Henrich, T.; Wittbrodt, J.; Loosli, F.; et al. TRIM25 has a dual function in the p53/Mdm2 circuit. Oncogene 2015, 34, 5729–5738. [Google Scholar] [CrossRef]
- Sakuma, M.; Akahira, J.; Suzuki, T.; Inoue, S.; Ito, K.; Moriya, T.; Sasano, H.; Okamura, K.; Yaegashi, N. Expression of estrogen-responsive finger protein (Efp) is associated with advanced disease in human epithelial ovarian cancer. Gynecol. Oncol. 2005, 99, 664–670. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, Y.; Zhang, C.; Yu, S.; Zhu, Q.; Hou, K.; Yan, B. TRIM25 blockade by RNA interference inhibited migration and invasion of gastric cancer cells through TGF-beta signaling. Sci. Rep. 2016, 6, 19070. [Google Scholar] [CrossRef]
- Qin, Y.; Cui, H.; Zhang, H. Overexpression of TRIM25 in Lung Cancer Regulates Tumor Cell Progression. Technol. Cancer Res. Treat. 2016, 15, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Xue, Y.; Dai, T.; Li, X.; Zheng, N. Tripartite motif containing 25 promotes proliferation and invasion of colorectal cancer cells through TGF-beta signaling. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasrullah, U.; Haeussler, K.; Biyanee, A.; Wittig, I.; Pfeilschifter, J.; Eberhardt, W. Identification of TRIM25 as a Negative Regulator of Caspase-2 Expression Reveals a Novel Target for Sensitizing Colon Carcinoma Cells to Intrinsic Apoptosis. Cells 2019, 8, 1622. https://doi.org/10.3390/cells8121622
Nasrullah U, Haeussler K, Biyanee A, Wittig I, Pfeilschifter J, Eberhardt W. Identification of TRIM25 as a Negative Regulator of Caspase-2 Expression Reveals a Novel Target for Sensitizing Colon Carcinoma Cells to Intrinsic Apoptosis. Cells. 2019; 8(12):1622. https://doi.org/10.3390/cells8121622
Chicago/Turabian StyleNasrullah, Usman, Kristina Haeussler, Abhiruchi Biyanee, Ilka Wittig, Josef Pfeilschifter, and Wolfgang Eberhardt. 2019. "Identification of TRIM25 as a Negative Regulator of Caspase-2 Expression Reveals a Novel Target for Sensitizing Colon Carcinoma Cells to Intrinsic Apoptosis" Cells 8, no. 12: 1622. https://doi.org/10.3390/cells8121622
APA StyleNasrullah, U., Haeussler, K., Biyanee, A., Wittig, I., Pfeilschifter, J., & Eberhardt, W. (2019). Identification of TRIM25 as a Negative Regulator of Caspase-2 Expression Reveals a Novel Target for Sensitizing Colon Carcinoma Cells to Intrinsic Apoptosis. Cells, 8(12), 1622. https://doi.org/10.3390/cells8121622