Relevance of Translation Initiation in Diffuse Glioma Biology and its Therapeutic Potential


 and
and
Abstract
1. Introduction
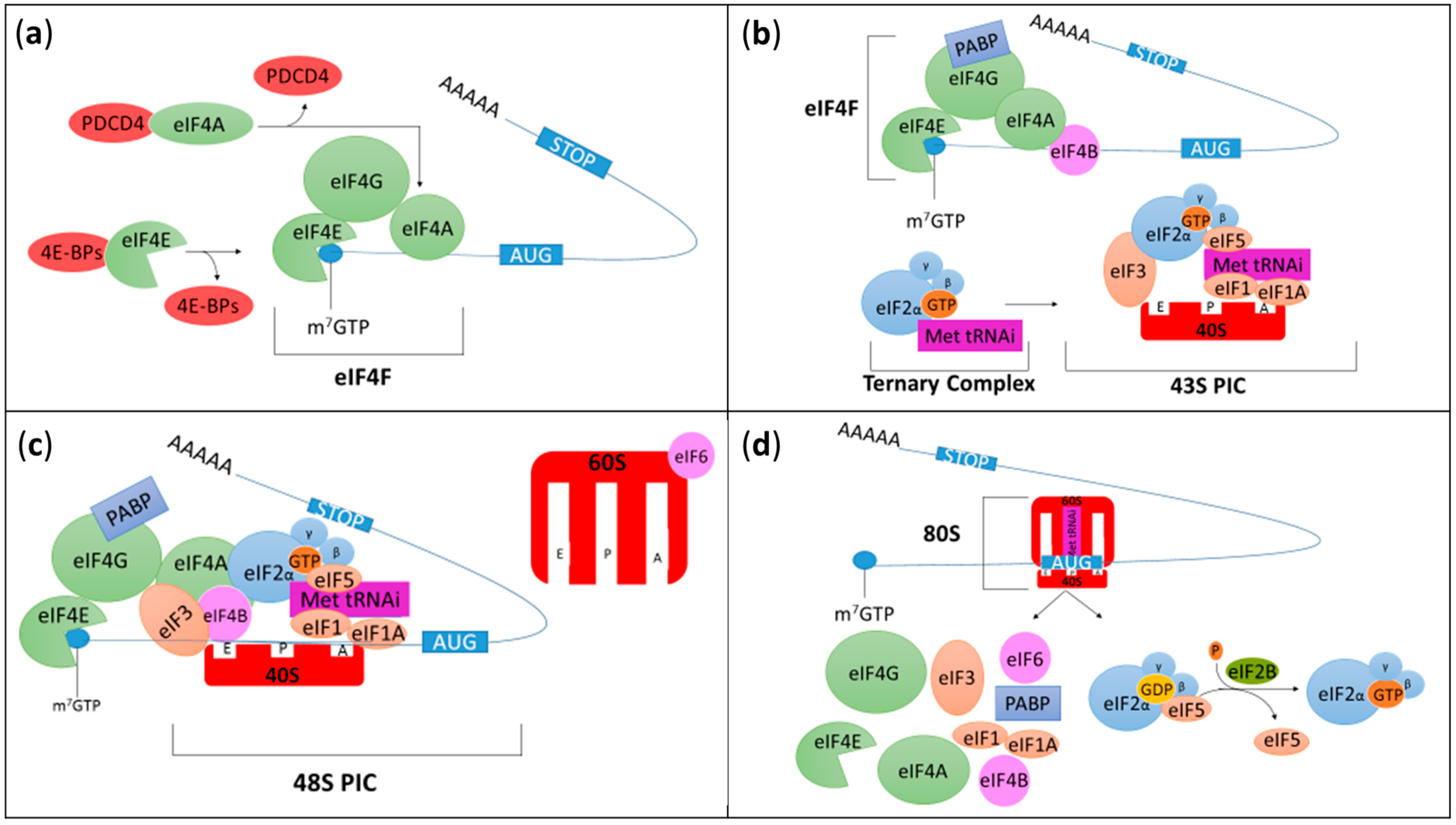
2. Overview on Translation Initiation
3. Cap-Dependent Initiation
3.1. The eIF4F Initiation Complex
3.1.1. eIF4E
3.1.2. eIF4G
3.1.3. eIF4A
3.2. The 43S Preinitiation Complex (PIC): eIF1, eIF1A, eIF3, and eIF5
3.2.1. eIF3
3.2.2. eIF1, eIF1A, eIF5, and eIF5B
3.3. Additional eIFs: eIF4B, eIF4H, and eIF6
3.3.1. eIF4B/eIF4H
3.3.2. eIF6
3.4. Ternary Complex (eIF2-GTP and the Initiator Methionyl-Transfer RNA)
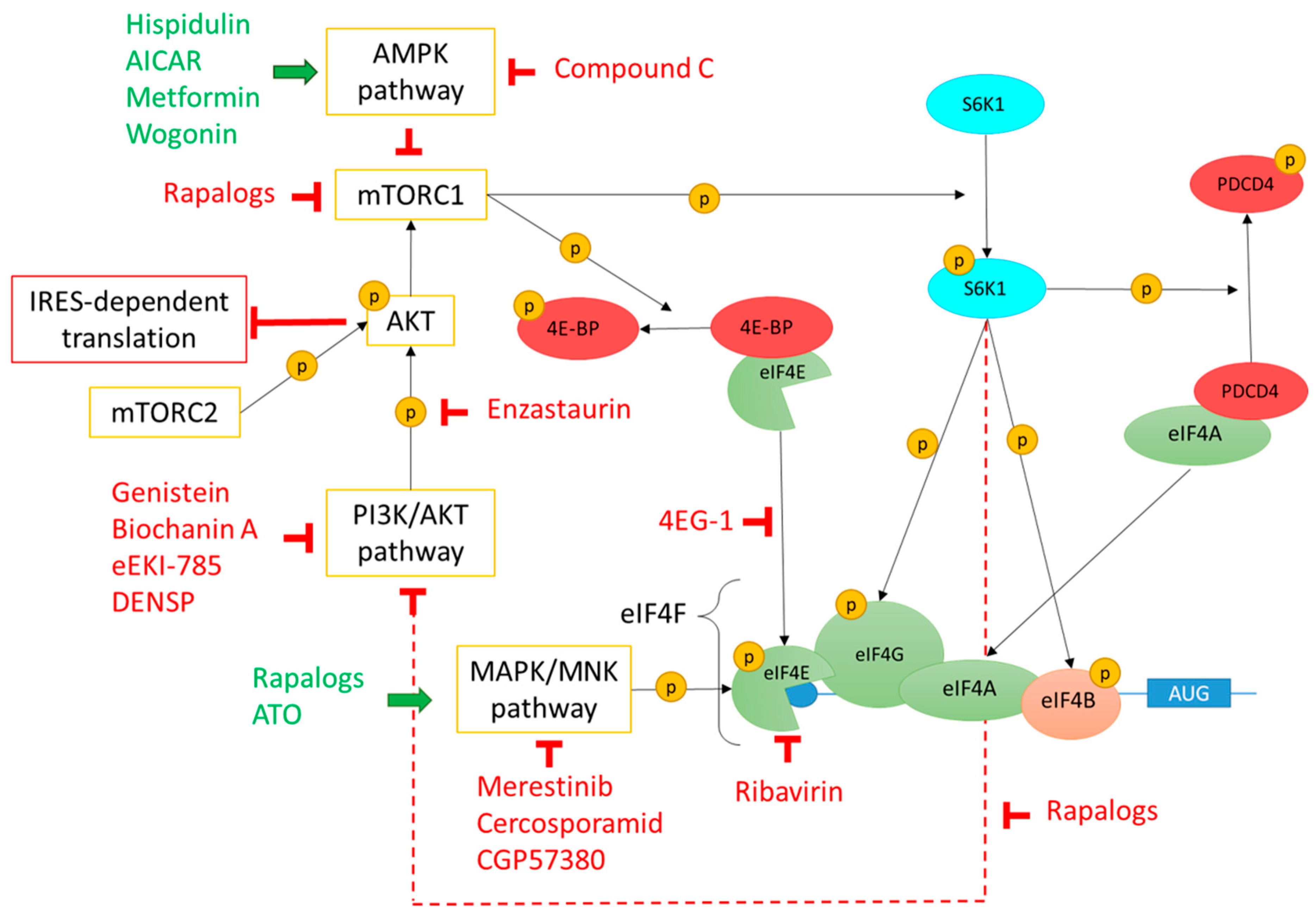
3.4.1. PI3K/Akt/mTOR, MAPK/MNK, and AMPK Regulating Pathways
3.4.2. PI3K/Akt/mTOR pathway
3.4.3. MAPK/MNK Pathway
3.4.4. AMPK Pathway
4. IRES-Dependent Initiation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, C.; Rodríguez Vázquez, L.; Martí, J. Glioblastoma Multiforme and Adult Neurogenesis in the Ventricular-Subventricular Zone: A Review. J. Cell. Physiol. 2017, 232, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Obacz, J.; Avril, T.; Le Reste, P.-J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma—From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017. [Google Scholar] [CrossRef]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2018. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Ding, Y.; Niemczyk, M.; Kudla, G.; Plotkin, J.B. Rate-limiting steps in yeast protein translation. Cell 2013. [Google Scholar] [CrossRef] [PubMed]
- Gusev, Y.; Bhuvaneshwar, K.; Song, L.; Zenklusen, J.-C.; Fine, H.; Madhavan, S. The REMBRANDT study, a large collection of genomic data from brain cancer patients. Sci. Data 2018. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Herdy, B.; Costa-Mattioli, M.; Gingras, A.-C.; Raught, B.; Sonenberg, N. Eukaryotic Translation Initiation Factor 4E Availability Controls the Switch between Cap-Dependent and Internal Ribosomal Entry Site-Mediated Translation. Mol. Cell. Biol. 2005, 25, 10556–10565. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Green, R. The Elongation, Termination, and Recycling Phases of Translation in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2012, 4, 13706. [Google Scholar] [CrossRef]
- Godet, A.-C.; David, F.; Hantelys, F.; Tatin, F.; Lacazette, E.; Garmy-Susini, B.; Prats, A.-C. IRES Trans-Acting Factors, Key Actors of the Stress Response. Int. J. Mol. Sci. 2019, 20, 924. [Google Scholar] [CrossRef]
- Shatsky, I.N.; Terenin, I.M.; Smirnova, V.V.; Andreev, D.E. Cap-Independent Translation: What’s in a Name? Trends Biochem. Sci. 2018, 43, 882–895. [Google Scholar] [CrossRef]
- Fan, W.; Wang, W.; Mao, X.; Chu, S.; Feng, J.; Xiao, D.; Zhou, J.; Fan, S. Elevated levels of p-Mnk1, p-eIF4E and p-p70S6K proteins are associated with tumor recurrence and poor prognosis in astrocytomas. J. Neurooncol. 2017, 131, 485–493. [Google Scholar] [CrossRef]
- Yang, S.X.; Hewitt, S.M.; Steinberg, S.M.; Liewehr, D.J.; Swain, S.M. Expression levels of eIF4E, VEGF, and cyclin D1, and correlation of eIF4E with VEGF and cyclin D1 in multi-tumor tissue microarray. Oncol. Rep. 2007, 17, 281–287. [Google Scholar] [CrossRef]
- Furic, L.; Rong, L.; Larsson, O.; Koumakpayi, I.H.; Yoshida, K.; Brueschke, A.; Petroulakis, E.; Robichaud, N.; Pollak, M.; Gabouryd, L.A.; et al. EIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA. 2010, 107, 14134–14139. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.; Cameron, A.; Jagus, R. Characterization of mammalian eIF4E-family members. Eur. J. Biochem. 2004, 271, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Jones, L.; Lowery-Norberg, M.; Fowler, M. Expression of eukaryotic initiation factor 4E in astrocytic tumors. Appl. Immunohistochem. Mol. Morphol. AIMM 2005, 13, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, E.; Peg, V.; Ortega-Aznar, A.; Martínez-Ricarte, F.; Camacho, J.; Hernández-Losa, J.; Ferreres Piñas, J.C.; Ramón y Cajal, S. peIF4E as an independent prognostic factor and a potential therapeutic target in diffuse infiltrating astrocytomas. Cancer Med. 2016, 5, 2501–2512. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Wu, Q.; Tian, T.; Li, L.; Guo, X.; Feng, Z.; Zhou, J.; Zhang, L.; Zhou, S.; Feng, G.; et al. The influence of SRPK1 on glioma apoptosis, metastasis, and angiogenesis through the PI3K/Akt signaling pathway under normoxia. Tumor Biol. 2015, 36, 6083–6093. [Google Scholar] [CrossRef]
- Kelly, N.J.; Varga, J.F.A.; Specker, E.J.; Romeo, C.M.; Coomber, B.L.; Uniacke, J. Hypoxia activates cadherin-22 synthesis via eIF4E2 to drive cancer cell migration, invasion and adhesion. Oncogene 2018, 37, 651–662. [Google Scholar] [CrossRef]
- Abdelfattah, N.; Rajamanickam, S.; Panneerdoss, S.; Timilsina, S.; Yadav, P.; Onyeagucha, B.C.; Garcia, M.; Vadlamudi, R.; Chen, Y.; Brenner, A.; et al. MiR-584-5p potentiates vincristine and radiation response by inducing spindle defects and DNA damage in medulloblastoma. Nat. Commun. 2018. [Google Scholar] [CrossRef]
- Cloninger, C.; Bernath, A.; Bashir, T.; Holmes, B.; Artinian, N.; Ruegg, T.; Anderson, L.; Masri, J.; Lichtenstein, A.; Gera, J. Inhibition of SAPK2/p38 Enhances Sensitivity to mTORC1 Inhibition by Blocking IRES-Mediated Translation Initiation in Glioblastoma. Mol. Cancer Ther. 2011, 10, 2244–2256. [Google Scholar] [CrossRef]
- Culjkovic, B.; Topisirovic, I.; Borden, K.L.B. Controlling Gene Expression through RNA Regulons: The Role of the Eukaryotic Translation Initiation Factor eIF4E. Cell Cycle 2007, 6, 65–69. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Ruggero, D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin. Cancer Res. 2010, 16, 4914–4920. [Google Scholar] [CrossRef]
- Sonenberg, N. Translation factors as effectors of cell growth and tumorigenesis. Curr. Opin. Cell Biol. 1993, 5, 955–960. [Google Scholar] [CrossRef]
- Rouschop, K.M.A.; Dubois, L.; Schaaf, M.B.E.; van den Beucken, T.; Lieuwes, N.; Keulers, T.G.H.; Savelkouls, K.G.M.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; et al. Deregulation of cap-dependent mRNA translation increases tumour radiosensitivity through reduction of the hypoxic fraction. Radiother. Oncol. 2011, 99, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Volpin, F.; Casaos, J.; Sesen, J.; Mangraviti, A.; Choi, J.; Gorelick, N.; Frikeche, J.; Lott, T.; Felder, R.; Scotland, S.J.; et al. Use of an anti-viral drug, Ribavirin, as an anti-glioblastoma therapeutic. Oncogene 2017, 36, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Shervington, A.; Munje, C.; Shervington, L. The Complexity of Identifying Cancer Stem Cell Biomarkers. Cancer Invest. 2013, 31, 404–411. [Google Scholar] [CrossRef]
- Ge, Y.; Zhou, F.; Chen, H.; Cui, C.; Liu, D.; Li, Q.; Yang, Z.; Wu, G.; Sun, S.; Gu, J.; et al. Sox2 is translationally activated by eukaryotic initiation factor 4E in human glioma-initiating cells. Biochem. Biophys. Res. Commun. 2010, 397, 711–717. [Google Scholar] [CrossRef]
- Goffart, N.; Lombard, A.; Lallemand, F.; Kroonen, J.; Nassen, J.; Di Valentin, E.; Berendsen, S.; Dedobbeleer, M.; Willems, E.; Robe, P.; et al. CXCL12 mediates glioblastoma resistance to radiotherapy in the subventricular zone. Neuro. Oncol. 2017, 19, 66–77. [Google Scholar] [CrossRef]
- Kroonen, J.; Nassen, J.; Boulanger, Y.-G.; Provenzano, F.; Capraro, V.; Bours, V.; Martin, D.; Deprez, M.; Robe, P.; Rogister, B. Human glioblastoma-initiating cells invade specifically the subventricular zones and olfactory bulbs of mice after striatal injection. Int. J. Cancer 2011, 129, 574–585. [Google Scholar] [CrossRef]
- Dostie, J.; Ferraiuolo, M.; Pause, A.; Adam, S.A.; Sonenberg, N. A novel shuttling protein, 4E-T, mediates the nuclear import of the mRNA 5’ cap-binding protein, eIF4E. EMBO J. 2000, 19, 3142–3156. [Google Scholar] [CrossRef]
- Nishimura, T.; Padamsi, Z.; Fakim, H.; Milette, S.; Dunham, W.H.; Gingras, A.-C.; Fabian, M.R. The eIF4E-Binding Protein 4E-T Is a Component of the mRNA Decay Machinery that Bridges the 5′ and 3′ Termini of Target mRNAs. Cell Rep. 2015, 11, 1425–1436. [Google Scholar] [CrossRef]
- Dobrikov, M.; Dobrikova, E.; Shveygert, M.; Gromeier, M. Phosphorylation of Eukaryotic Translation Initiation Factor 4G1 (eIF4G1) by Protein Kinase C Regulates eIF4G1 Binding to Mnk1. Mol. Cell. Biol. 2011, 31, 2947–2959. [Google Scholar] [CrossRef] [PubMed]
- Hundsdoerfer, P.; Thoma, C.; Hentze, M.W. Eukaryotic translation initiation factor 4GI and p97 promote cellular internal ribosome entry sequence-driven translation. Proc. Natl. Acad. Sci. USA 2005, 102, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Pyronnet, S.; Imataka, H.; Gingras, A.C.; Fukunaga, R.; Hunter, T.; Sonenberg, N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits Mnk1 to phosphorylate eIF4E. EMBO J. 1999, 18, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Kahvejian, A.; Svitkin, Y.V.; Sukarieh, R.; M’Boutchou, M.-N.; Sonenberg, N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005, 19, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Cargnello, M.; Topisirovic, I.; Pelletier, J. Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol. 2016, 26, 918–933. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, C.; Li, X.-J.; Liu, Q.; Wanggou, S. Anti-Cancer Effect of Cap-Translation Inhibitor 4EGI-1 in Human Glioma U87 Cells: Involvement of Mitochondrial Dysfunction and ER Stress. Cell. Physiol. Biochem. 2016, 40, 1013–1028. [Google Scholar] [CrossRef]
- Yang, X.; Dong, Q.-F.; Li, L.-W.; Huo, J.-L.; Li, P.-Q.; Fei, Z.; Zhen, H.-N. The cap-translation inhibitor 4EGI-1 induces mitochondrial dysfunction via regulation of mitochondrial dynamic proteins in human glioma U251 cells. Neurochem. Int. 2015, 90, 98–106. [Google Scholar] [CrossRef]
- Dubois, L.; Magagnin, M.G.; Cleven, A.H.G.; Weppler, S.A.; Grenacher, B.; Landuyt, W.; Lieuwes, N.; Lambin, P.; Gorr, T.A.; Koritzinsky, M.; et al. Inhibition of 4E-BP1 Sensitizes U87 Glioblastoma Xenograft Tumors to Irradiation by Decreasing Hypoxia Tolerance. Int. J. Radiat. Oncol. 2009, 73, 1219–1227. [Google Scholar] [CrossRef]
- Raza, F.; Waldron, J.A.; Quesne, J.L. Translational dysregulation in cancer: eIF4A isoforms and sequence determinants of eIF4A dependence. Biochem. Soc. Trans. 2015, 43, 1227–1233. [Google Scholar] [CrossRef]
- Parsyan, A.; Svitkin, Y.; Shahbazian, D.; Gkogkas, C.; Lasko, P.; Merrick, W.C.; Sonenberg, N. MRNA helicases: The tacticians of translational control. Nat. Rev. Mol. Cell Biol. 2011, 12, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Shaoyan, X.; Juanjuan, Y.; Yalan, T.; Ping, H.; Jianzhong, L.; Qinian, W. Downregulation of EIF4A2 in Non–Small-Cell Lung Cancer Associates with Poor Prognosis. Clin. Lung Cancer 2013, 14, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.X.; Wu, Q.N.; Zhang, Y.; Li, Y.Y.; Liao, D.Z.; Hou, J.H.; Fu, J.; Zeng, M.S.; Yun, J.P.; Wu, Q.L.; et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivotumor growth. Breast Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.; Jansen, A.P.; Baker, A.R.; Hegamyer, G.; Hagan, J.P.; Colburn, N.H. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res. 2008, 68, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-K.; Baker, A.R.; Young, M.R.; Colburn, N.H. Tumor suppressor PDCD4 inhibits NF-κB-dependent transcription in human glioblastoma cells by direct interaction with p65. Carcinogenesis 2014, 35, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-S.; Cho, M.-H.; Zakowicz, H.; Hegamyer, G.; Sonenberg, N.; Colburn, N.H. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol. Cell. Biol. 2004, 24, 3894–3906. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, P.; Zhou, C.; Li, J.; Wang, Q.; Zhu, F.; Ma, C.; Sun, W.; Zhang, L. Frequent loss of PDCD4 expression in human glioma: Possible role in the tumorigenesis of glioma. Oncol. Rep. 2007, 17, 123–128. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Yang, H.-S. The role of Pdcd4 in tumour suppression and protein translation. Biol. Cell 2018, 110, 169–177. [Google Scholar] [CrossRef]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 Is an Antiapoptotic Factor in Human Glioblastoma Cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Ma, Q.; Huang, J.; Xiong, Y.; Yang, X.; Han, R.; Zhu, W. MicroRNA-96 Regulates Apoptosis by Targeting PDCD4 in Human Glioma Cells. Technol. Cancer Res. Treat. 2017, 16, 92–98. [Google Scholar] [CrossRef]
- Bordeleau, M.E.; Matthews, J.; Wojnar, J.M.; Lindqvist, L.; Novac, O.; Jankowsky, E.; Sonenberg, N.; Northcote, P.; Teesdale-Spittle, P.; Pelletier, J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc. Natl. Acad. Sci. USA. 2005, 102, 10460–10465. [Google Scholar] [CrossRef] [PubMed]
- Low, W.K.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Merrick, W.C.; Romo, D.; Liu, J.O. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 2005, 20, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Kolupaeva, V.G. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002, 16, 2906–2922. [Google Scholar] [CrossRef] [PubMed]
- des Georges, A.; Dhote, V.; Kuhn, L.; Hellen, C.U.T.; Pestova, T.V.; Frank, J.; Hashem, Y. Structure of mammalian eIF3 in the context of the 43S preinitiation complex. Nature 2015, 525, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, E.; Luna, R.E.; Nagata, T.; Martin-Marcos, P.; Hiraishi, H.; Singh, C.R.; Erzberger, J.P.; Zhang, F.; Arthanari, H.; Morris, J.; et al. Molecular Landscape of the Ribosome Pre-initiation Complex during mRNA Scanning: Structural Role for eIF3c and Its Control by eIF5. Cell Rep. 2017, 18, 2651–2663. [Google Scholar] [CrossRef] [PubMed]
- Sokabe, M.; Fraser, C.S.; Hershey, J.W.B. The human translation initiation multi-factor complex promotes methionyl-tRNA i binding to the 40S ribosomal subunit. Nucleic Acids Res. 2012, 40, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Villa, N.; Do, A.; Hershey, J.W.B.; Fraser, C.S. Human eukaryotic initiation factor 4G (eIF4G) protein binds to eIF3c, -d, and -e to promote mRNA recruitment to the ribosome. J. Biol. Chem. 2013, 288, 32932–32940. [Google Scholar] [CrossRef]
- Hershey, J.W.B. The role of eIF3 and its individual subunits in cancer. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 792–800. [Google Scholar] [CrossRef]
- Emmanuel, R.; Weinstein, S.; Landesman-Milo, D.; Peer, D. eIF3c: A potential therapeutic target for cancer. Cancer Lett. 2013, 336, 158–166. [Google Scholar] [CrossRef]
- Fan, Y.; Guo, Y. Knockdown of eIF3D inhibits breast cancer cell proliferation and invasion through suppressing the Wnt/β-catenin signaling pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 10420–10427. [Google Scholar]
- Li, Z.; Lin, S.; Jiang, T.; Wang, J.; Lu, H.; Tang, H.; Teng, M.; Fan, J. Overexpression of eIF3e is correlated with colon tumor development and poor prognosis. Int. J. Clin. Exp. Pathol. 2014, 7, 6462–6474. [Google Scholar] [PubMed]
- Spilka, R.; Ernst, C.; Bergler, H.; Rainer, J.; Flechsig, S.; Vogetseder, A.; Lederer, E.; Benesch, M.; Brunner, A.; Geley, S.; et al. eIF3a is over-expressed in urinary bladder cancer and influences its phenotype independent of translation initiation. Cell. Oncol. 2014, 37, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ru, Y.; Sanchez-Carbayo, M.; Wang, X.; Kieft, J.S.; Theodorescu, D. Translation Initiation Factor eIF3b Expression in Human Cancer and Its Role in Tumor Growth and Lung Colonization. Clin. Cancer Res. 2013, 19, 2850–2860. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.-C.; Wang, N.; Chang, Y.-Z.; Zhang, K.-N.; Li, J.-J.; Niu, J.-J.; Wu, F.; Liu, Y.-Q.; Wang, Y.-Z. Systematically profiling the expression of eIF3 subunits in glioma reveals the expression of eIF3i has prognostic value in IDH-mutant lower grade glioma. Cancer Cell Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- HAO, J.; LIANG, C.; JIAO, B. Eukaryotic translation initiation factor 3, subunit C is overexpressed and promotes cell proliferation in human glioma U-87 MG cells. Oncol. Lett. 2015, 9, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ding, X.; Zhou, C.; Zhang, Y.; Xu, M.; Zhang, C.; Xu, L. Knockdown of eukaryotic translation initiation factors 3B (EIF3B) inhibits proliferation and promotes apoptosis in glioblastoma cells. Neurol. Sci. 2012, 33, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhou, C.; Liang, H.; Wang, X.; Xu, L. RNAi-Mediated Silencing of EIF3D Alleviates Proliferation and Migration of Glioma U251 and U87MG Cells. Chem. Biol. Drug Des. 2015, 86, 715–722. [Google Scholar] [CrossRef]
- Sesen, J.; Cammas, A.; Scotland, S.; Elefterion, B.; Lemarié, A.; Millevoi, S.; Mathew, L.; Seva, C.; Toulas, C.; Moyal, E.; et al. Int6/eIF3e Is Essential for Proliferation and Survival of Human Glioblastoma Cells. Int. J. Mol. Sci. 2014, 15, 2172–2190. [Google Scholar] [CrossRef]
- Lee, A.S.Y.; Kranzusch, P.J.; Cate, J.H.D. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 2015, 522, 111–114. [Google Scholar] [CrossRef]
- Passmore, L.A.; Schmeing, T.M.; Maag, D.; Applefield, D.J.; Acker, M.G.; Algire, M.A.; Lorsch, J.R.; Ramakrishnan, V. The Eukaryotic Translation Initiation Factors eIF1 and eIF1A Induce an Open Conformation of the 40S Ribosome. Mol. Cell 2007, 26, 41–50. [Google Scholar] [CrossRef]
- Yu, J.; Marintchev, A. Comparative sequence and structure analysis of eIF1A and eIF1AD. BMC Struct. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Vanden Dungen, K.; Bressler, K.R.; Fredriksen, M.; Khandige Sharma, D.; Balasingam, N.; Thakor, N. Eukaryotic initiation factor 5B (eIF5B) provides a critical cell survival switch to glioblastoma cells via regulation of apoptosis. Cell Death Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Andreou, A.Z.; Harms, U.; Klostermeier, D. eIF4B stimulates eIF4A ATPase and unwinding activities by direct interaction through its 7-repeats region. RNA Biol. 2017, 14, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Rozovsky, N.; Butterworth, A.C.; Moore, M.J. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA 2008, 14, 2136–2148. [Google Scholar] [CrossRef]
- Shahbazian, D.; Parsyan, A.; Petroulakis, E.; Hershey, J.W.B.; Sonenberg, N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle 2010, 9, 4106–4109. [Google Scholar] [CrossRef]
- Vaysse, C.; Philippe, C.; Martineau, Y.; Quelen, C.; Hieblot, C.; Renaud, C.; Nicaise, Y.; Desquesnes, A.; Pannese, M.; Filleron, T.; et al. Key contribution of eIF4H-mediated translational control in tumor promotion. Oncotarget 2015, 6, 39924–39940. [Google Scholar] [CrossRef]
- Wang, Y.; Begley, M.; Li, Q.; Huang, H.-T.; Lako, A.; Eck, M.J.; Gray, N.S.; Mitchison, T.J.; Cantley, L.C.; Zhao, J.J. Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proc. Natl. Acad. Sci. USA 2016, 113, 9810–9815. [Google Scholar] [CrossRef]
- Kroczynska, B.; Kaur, S.; Katsoulidis, E.; Majchrzak-Kita, B.; Sassano, A.; Kozma, S.C.; Fish, E.N.; Platanias, L.C. Interferon-Dependent Engagement of Eukaryotic Initiation Factor 4B via S6 Kinase (S6K)- and Ribosomal Protein S6K-Mediated Signals. Mol. Cell. Biol. 2009, 29, 2865–2875. [Google Scholar] [CrossRef][Green Version]
- Zhu, W.; Li, G.X.; Chen, H.L.; Liu, X.Y. The role of eukaryotic translation initiation factor 6 in tumors. Oncol. Lett. 2017, 14, 3–9. [Google Scholar] [CrossRef]
- Sanvito, F.; Piatti, S.; Villa, A.; Bossi, M.; Lucchini, G.; Marchisio, P.C.; Biffo, S. The β4 Integrin Interactor p27 BBP/eIF6 Is an Essential Nuclear Matrix Protein Involved in 60S Ribosomal Subunit Assembly. J. Cell Biol. 1999, 144, 823–838. [Google Scholar] [CrossRef]
- Saito, K.; Iizuka, Y.; Ohta, S.; Takahashi, S.; Nakamura, K.; Saya, H.; Yoshida, K.; Kawakami, Y.; Toda, M. Functional analysis of a novel glioma antigen, EFTUD1. Neuro. Oncol. 2014, 16, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.D.; Zhou, Y.; Mohammad-Qureshi, S.S.; Bennett, D.; Pavitt, G.D. eIF2B promotes eIF5 dissociation from eIF2*GDP to facilitate guanine nucleotide exchange for translation initiation. Genes Dev. 2013, 27, 2696–2707. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hinnebusch, A.G. Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2. Mol. Cell. Biol. 1996, 16, 6603–6616. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fabian, J.R.; Kimball, S.R.; Heinzinger, N.K.; Jefferson, L.S. Subunit Assembly and Guanine Nucleotide Exchange Activity of Eukaryotic Initiation Factor-2B Expressed in Sf9 Cells. J. Biol. Chem. 1997, 272, 12359–12365. [Google Scholar] [CrossRef] [PubMed]
- Fabian, J.R.; Kimball, S.R.; Jefferson, L.S. Reconstitution and Purification of Eukaryotic Initiation Factor 2B (eIF2B) Expressed in Sf21 Insect Cells. Protein Expr. Purif. 1998, 13, 16–22. [Google Scholar] [CrossRef]
- Gordiyenko, Y.; Schmidt, C.; Jennings, M.D.; Matak-Vinkovic, D.; Pavitt, G.D.; Robinson, C.V. eIF2B is a decameric guanine nucleotide exchange factor with a γ2ε2 tetrameric core. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Gallagher, J.W.; Kubica, N.; Kimball, S.R.; Jefferson, L.S. Reduced Eukaryotic Initiation Factor 2Bε-Subunit Expression Suppresses the Transformed Phenotype of Cells Overexpressing the Protein. Cancer Res. 2008, 68, 8752–8760. [Google Scholar] [CrossRef]
- Yatime, L.; Mechulam, Y.; Blanquet, S.; Schmitt, E. Structure of an archaeal heterotrimeric initiation factor 2 reveals a nucleotide state between the GTP and the GDP states. Proc. Natl. Acad. Sci. USA 2007, 104, 18445–18450. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Wek, R.C. Phosphorylation of the α-Subunit of the Eukaryotic Initiation Factor-2 (eIF2α) Reduces Protein Synthesis and Enhances Apoptosis in Response to Proteasome Inhibition. J. Biol. Chem. 2005, 280, 14189–14202. [Google Scholar] [CrossRef]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Haapa-Paananen, S.; Chen, P.; Hellström, K.; Kohonen, P.; Hautaniemi, S.; Kallioniemi, O.; Perälä, M. Functional Profiling of Precursor MicroRNAs Identifies MicroRNAs Essential for Glioma Proliferation. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, I.; Eizenbach, M.; Sajman, J.; Ben-Bassat, H.; Levitzki, A. A cellular screening assay to test the ability of PKR to induce cell death in mammalian cells. Mol. Ther. 2005, 12, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Shir, A.; Ogris, M.; Wagner, E.; Levitzki, A. EGF Receptor-Targeted Synthetic Double-Stranded RNA Eliminates Glioblastoma, Breast Cancer, and Adenocarcinoma Tumors in Mice. PLoS Med. 2005. [Google Scholar] [CrossRef] [PubMed]
- Chaveroux, C.; Bruhat, A.; Carraro, V.; Jousse, C.; Averous, J.; Maurin, A.-C.; Parry, L.; Mesclon, F.; Muranishi, Y.; Cordelier, P.; et al. Regulating the expression of therapeutic transgenes by controlled intake of dietary essential amino acids. Nat. Biotechnol. 2016, 34, 746–751. [Google Scholar] [CrossRef]
- Adam, I.; Dewi, D.L.; Mooiweer, J.; Sadik, A.; Mohapatra, S.R.; Berdel, B.; Keil, M.; Sonner, J.K.; Thedieck, K.; Rose, A.J.; et al. Upregulation of tryptophanyl-tRNA synthethase adapts human cancer cells to nutritional stress caused by tryptophan degradation. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef]
- Harvey, R.F.; Willis, A.E. Post-transcriptional control of stress responses in cancer. Curr. Opin. Genet. Dev. 2018, 48, 30–35. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Peñaranda Fajardo, N.M.; Meijer, C.; Kruyt, F.A.E. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Tsai, C.-F.; Yeh, W.-L.; Huang, S.M.; Tan, T.-W.; Lu, D.-Y. Wogonin Induces Reactive Oxygen Species Production and Cell Apoptosis in Human Glioma Cancer Cells. Int. J. Mol. Sci. 2012, 13, 9877–9892. [Google Scholar] [CrossRef]
- Lu, D.-Y.; Chang, C.-S.; Yeh, W.-L.; Tang, C.-H.; Cheung, C.-W.; Leung, Y.-M.; Liu, J.-F.; Wong, K.-L. The novel phloroglucinol derivative BFP induces apoptosis of glioma cancer through reactive oxygen species and endoplasmic reticulum stress pathways. Phytomedicine 2012, 19, 1093–1100. [Google Scholar] [CrossRef]
- Tungkum, W.; Jumnongprakhon, P.; Tocharus, C.; Govitrapong, P.; Tocharus, J. Melatonin suppresses methamphetamine-triggered endoplasmic reticulum stress in C6 cells glioma cell lines. J. Toxicol. Sci. 2017, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Gabrusiewicz, K.; Szczepankiewicz, A.A.; Kaminska, B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine A-induced cell death. Oncogene 2013, 32, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Cheng, C.; Hackett, C.; Feldman, M.; Houseman, B.T.; Nicolaides, T.; Haas-Kogan, D.; James, C.D.; Oakes, S.A.; Debnath, J.; et al. Akt and Autophagy Cooperate to Promote Survival of Drug-Resistant Glioma. Sci. Signal. 2010, 3, 81. [Google Scholar] [CrossRef] [PubMed]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Loria, R.M.; Park, M.A.; Yacoub, A.; Dent, P.; Graf, M.R. The neuro-steroid, 5-androstene 3β,17α diol; induces endoplasmic reticulum stress and autophagy through PERK/eIF2α signaling in malignant glioma cells and transformed fibroblasts. Int. J. Biochem. Cell Biol. 2010, 42, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-T.; Huang, C.-Y.; Lu, I.-C.; Gean, P.-W. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro. Oncol. 2013, 15, 1127–1141. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Wang, Z.; Zhang, R.; Gong, X. Bufalin Induces the Interplay between Apoptosis and Autophagy in Glioma Cells through Endoplasmic Reticulum Stress. Int. J. Biol. Sci. 2014, 10, 212–224. [Google Scholar] [CrossRef]
- Anderson, K.C. The 39th David, A. Karnofsky Lecture: Bench-to-bedside translation of targeted therapies in multiple myeloma. J. Clin. Oncol. 2012, 30, 445–452. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Hatzidaki, E.; Stathakis, N.E.; Koukoulis, G.K.; Papandreou, C.N. Bortezomib Downregulates MGMT Expression in T98G Glioblastoma Cells. Cell. Mol. Neurobiol. 2013, 33, 313–318. [Google Scholar] [CrossRef]
- Vilas-Boas, F.d.A.S.; da Silva, A.M.; de Sousa, L.P.; Lima, K.M.; Vago, J.P.; Bittencourt, L.F.F.; Dantas, A.E.; Gomes, D.A.; Vilela, M.C.; Teixeira, M.M.; et al. Impairment of stress granule assembly via inhibition of the eIF2alpha phosphorylation sensitizes glioma cells to chemotherapeutic agents. J. Neurooncol. 2016, 127, 253–260. [Google Scholar]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro. Oncol. 2012, 14, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Gaut, D.; Hu, K.; Yan, H.; Yin, D.; Koeffler, H.P. Dual targeting of glioblastoma multiforme with a proteasome inhibitor (Velcade) and a phosphatidylinositol 3-kinase inhibitor (ZSTK474). Int. J. Oncol. 2014, 44, 557–562. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, S.; Lee, D.; Ho, A.S.W.; Pu, J.K.S.; Zhang, X.Q.; Lee, N.P.; Day, P.J.R.; Lui, W.M.; Fung, C.F.; Leung, G.K.K. Inhibition of prolyl 4-hydroxylase, beta polypeptide (P4HB) attenuates temozolomide resistance in malignant glioma via the endoplasmic reticulum stress response (ERSR) pathways. Neuro. Oncol. 2013, 15, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; Lee, J.; Landon, K.A.; Benavides-Serrato, A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Mechanistic Target of Rapamycin (mTOR) Inhibition Synergizes with Reduced Internal Ribosome Entry Site (IRES)-mediated Translation of Cyclin D1 and c-MYC mRNAs to Treat Glioblastoma. J. Biol. Chem. 2016, 291, 14146–14159. [Google Scholar] [CrossRef]
- Martin, J.; Masri, J.; Cloninger, C.; Holmes, B.; Artinian, N.; Funk, A.; Ruegg, T.; Anderson, L.; Bashir, T.; Bernath, A.; et al. Phosphomimetic Substitution of Heterogeneous Nuclear Ribonucleoprotein A1 at Serine 199 Abolishes AKT-dependent Internal Ribosome Entry Site-transacting Factor (ITAF) Function via Effects on Strand Annealing and Results in Mammalian Target of Rapamycin Complex 1 (mTORC1) Inhibitor Sensitivity. J. Biol. Chem. 2011, 286, 16402–16413. [Google Scholar]
- Ueda, T.; Sasaki, M.; Elia, A.J.; Chio, I.I.C.; Hamada, K.; Fukunaga, R.; Mak, T.W. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc. Natl. Acad. Sci. USA 2010, 107, 13984–13990. [Google Scholar] [CrossRef]
- Bell, J.B.; Eckerdt, F.; Dhruv, H.D.; Finlay, D.; Peng, S.; Kim, S.; Kroczynska, B.; Beauchamp, E.M.; Alley, K.; Clymer, J.; et al. Differential Response of Glioma Stem Cells to Arsenic Trioxide Therapy Is Regulated by MNK1 and mRNA Translation. Mol. Cancer Res. 2018, 16, 32–46. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Grzmil, M.; Huber, R.M.; Hess, D.; Frank, S.; Hynx, D.; Moncayo, G.; Klein, D.; Merlo, A.; Hemmings, B.A. MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J. Clin. Investig. 2014, 124, 742–754. [Google Scholar] [CrossRef]
- Laderoute, K.R.; Calaoagan, J.M.; Chao, W.; Dinh, D.; Denko, N.; Duellman, S.; Kalra, J.; Liu, X.; Papandreou, I.; Sambucetti, L.; et al. 5′-AMP-activated Protein Kinase (AMPK) Supports the Growth of Aggressive Experimental Human Breast Cancer Tumors. J. Biol. Chem. 2014, 289, 22850–22864. [Google Scholar] [CrossRef]
- Masri, J.; Bernath, A.; Martin, J.; Jo, O.D.; Vartanian, R.; Funk, A.; Gera, J. mTORC2 Activity Is Elevated in Gliomas and Promotes Growth and Cell Motility via Overexpression of Rictor. Cancer Res. 2007, 67, 11712–11720. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A Cellular Regulator Interface in Health and Disease. Cells 2019. [Google Scholar] [CrossRef] [PubMed]
- Duzgun, Z.; Eroglu, Z.; Biray Avci, C. Role of mtomTOR in glioblastoma. Gene 2016, 575, 187–190. [Google Scholar] [CrossRef]
- Gulati, N.; Karsy, M.; Albert, L.; Murali, R.; Jhanwar-Uniyal, M. Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int. J. Oncol. 2009, 35, 731–740. [Google Scholar]
- Urbanska, M.; Gozdz, A.; Swiech, L.J.; Jaworski, J. Mammalian Target of Rapamycin Complex 1 (mTORC1) and 2 (mTORC2) Control the Dendritic Arbor Morphology of Hippocampal Neurons. J. Biol. Chem. 2012, 287, 30240–30256. [Google Scholar] [CrossRef]
- Korkolopoulou, P.; Levidou, G.; El-Habr, E.A.; Piperi, C.; Adamopoulos, C.; Samaras, V.; Boviatsis, E.; Thymara, I.; Trigka, E.-A.; Sakellariou, S.; et al. Phosphorylated 4E-binding protein 1 (p-4E-BP1): A novel prognostic marker in human astrocytomas. Histopathology 2012, 61, 293–305. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Amin, A.G.; Cooper, J.B.; Das, K.; Schmidt, M.H.; Murali, R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv. Biol. Regul. 2017, 64, 39–48. [Google Scholar] [CrossRef]
- Dumstorf, C.A.; Konicek, B.W.; McNulty, A.M.; Parsons, S.H.; Furic, L.; Sonenberg, N.; Graff, J.R. Modulation of 4E-BP1 Function as a Critical Determinant of Enzastaurin-Induced Apoptosis. Mol. Cancer Ther. 2010, 9, 3158–3163. [Google Scholar] [CrossRef]
- Jiang, R.; Choi, W.; Hu, L.; Gerner, E.W.; Hamilton, S.R.; Zhang, W. Activation of polyamine catabolism by N1, N11-diethylnorspermine alters the cellular localization of mTOR and downregulates mTOR protein level in glioblastoma cells. Cancer Biol. Ther. 2007, 6, 1644–1648. [Google Scholar] [CrossRef][Green Version]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor Activity of Rapamycin in a Phase I Trial for Patients with Recurrent PTEN-Deficient Glioblastoma. PLoS Med. 2008. [Google Scholar] [CrossRef]
- Puli, S.; Jain, A.; Lai, J.C.K.; Bhushan, A. Effect of Combination Treatment of Rapamycin and Isoflavones on mTOR Pathway in Human Glioblastoma (U87) Cells. Neurochem. Res. 2010, 35, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.D.; Mladek, A.C.; Lamont, J.D.; Goble, J.M.; Erlichman, C.; James, C.D.; Sarkaria, J.N. Disruption of Parallel and Converging Signaling Pathways Contributes to the Synergistic Antitumor Effects of Simultaneous mTOR and EGFR Inhibition in GBM Cells. Neoplasia 2005, 7, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.B.; Eckerdt, F.D.; Alley, K.; Magnusson, L.P.; Hussain, H.; Bi, Y.; Arslan, A.D.; Clymer, J.; Alvarez, A.A.; Goldman, S.; et al. MNK Inhibition Disrupts Mesenchymal Glioma Stem Cells and Prolongs Survival in a Mouse Model of Glioblastoma. Mol. Cancer Res. 2016, 14, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef]
- Brown, M.C.; Dobrikov, M.I.; Gromeier, M. Mitogen-Activated Protein Kinase-Interacting Kinase Regulates mTOR/AKT Signaling and Controls the Serine/Arginine-Rich Protein Kinase-Responsive Type 1 Internal Ribosome Entry Site-Mediated Translation and Viral Oncolysis. J. Virol. 2014, 88, 13149–13160. [Google Scholar] [CrossRef]
- Zheng, J.; Li, J.; Xu, L.; Xie, G.; Wen, Q.; Luo, J.; Li, D.; Huang, D.; Fan, S. Phosphorylated Mnk1 and eIF4E Are Associated with Lymph Node Metastasis and Poor Prognosis of Nasopharyngeal Carcinoma. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Lineham, E.; Spencer, J.; Morley, S.J. Dual abrogation of MNK and mTOR: A novel therapeutic approach for the treatment of aggressive cancers. Future Med. Chem. 2017, 9, 1539–1555. [Google Scholar] [CrossRef]
- Grzmil, M.; Morin, P.; Lino, M.M.; Merlo, A.; Frank, S.; Wang, Y.; Moncayo, G.; Hemmings, B.A. MAP Kinase-Interacting Kinase 1 Regulates SMAD2-Dependent TGF- Signaling Pathway in Human Glioblastoma. Cancer Res. 2011, 71, 2392–2402. [Google Scholar] [CrossRef]
- Grzmil, M.; Seebacher, J.; Hess, D.; Behe, M.; Schibli, R.; Moncayo, G.; Frank, S.; Hemmings, B.A. Inhibition of MNK pathways enhances cancer cell response to chemotherapy with temozolomide and targeted radionuclide therapy. Cell. Signal. 2016, 28, 1412–1421. [Google Scholar] [CrossRef]
- Ng, T.L.; Leprivier, G.; Robertson, M.D.; Chow, C.; Martin, M.J.; Laderoute, K.R.; Davicioni, E.; Triche, T.J.; Sorensen, P.H.B. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012, 19, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; McTiernan, A.; Wactawski-Wende, J.; Manson, J.E.; Aragaki, A.K.; Rohan, T.; Ipp, E.; Kaklamani, V.G.; Vitolins, M.; Wallace, R.; et al. Diabetes, Metformin, and Breast Cancer in Postmenopausal Women. J. Clin. Oncol. 2012, 30, 2844–2852. [Google Scholar] [CrossRef] [PubMed]
- Tai, M.C.; Tsang, S.Y.; Chang, L.Y.F.; Xue, H. Therapeutic potential of wogonin: A naturally occurring flavonoid. CNS Drug Rev. 2005, 11, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Van Den Neste, E.; Cazin, B.; Janssens, A.; González-Barca, E.; Terol, M.J.; Levy, V.; Pérez de Oteyza, J.; Zachee, P.; Saunders, A.; de Frias, M.; et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): A multicenter phase I/II study. Cancer Chemother. Pharmacol. 2013, 71, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-J.; Zheng, Z.-J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced Risk of Colorectal Cancer With Metformin Therapy in Patients With Type 2 Diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.-J.; Watson, A.D.; Phelps, M.; et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef]
- Lee, D.-H.; Lee, T.H.; Jung, C.H.; Kim, Y.-H. Wogonin induces apoptosis by activating the AMPK and p53 signaling pathways in human glioblastoma cells. Cell. Signal. 2012, 24, 2216–2225. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Hung, C.-M.; Tsai, J.-C.; Lee, J.-C.; Chen, Y.-L.S.; Wei, C.-W.; Kao, J.-Y.; Way, T.-D. Hispidulin Potently Inhibits Human Glioblastoma Multiforme Cells through Activation of AMP-Activated Protein Kinase (AMPK). J. Agric. Food Chem. 2010, 58, 9511–9517. [Google Scholar] [CrossRef]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.-T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Cohen-Jonathan Moyal, E.; et al. Metformin Inhibits Growth of Human Glioblastoma Cells and Enhances Therapeutic Response. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK Inhibitor Compound C Is a Potent AMPK-Independent Antiglioma Agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.L.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, L.; Misirkic, M.; Janjetovic, K.; Harhaji-Trajkovic, L.; Prica, M.; Stevanovic, D.; Isenovic, E.; Sudar, E.; Sumarac-Dumanovic, M.; Micic, D.; et al. AMP-activated protein kinase-dependent and -independent mechanisms underlying in vitro antiglioma action of compound C. Biochem. Pharmacol. 2009, 77, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Ogura, T.; Kishimoto, A.; Minegishi, Y.; Nakajima, N.; Miyazaki, M.; Esumi, H. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene 2002, 21, 6082–6090. [Google Scholar] [CrossRef]
- Yang, Q.; Sarnow, P. Location of the internal ribosome entry site in the 5’ non-coding region of the immunoglobulin heavy-chain binding protein (BiP) mRNA: Evidence for specific RNA-protein interactions. Nucleic Acids Res. 1997, 25, 2800–2807. [Google Scholar] [CrossRef]
- Blau, L.; Knirsh, R.; Ben-Dror, I.; Oren, S.; Kuphal, S.; Hau, P.; Proescholdt, M.; Bosserhoff, A.-K.; Vardimon, L. Aberrant expression of c-Jun in glioblastoma by internal ribosome entry site (IRES)-mediated translational activation. Proc. Natl. Acad. Sci. USA 2012, 109, 2875–2884. [Google Scholar] [CrossRef]
- Webb, T.E.; Hughes, A.; Smalley, D.S.; Spriggs, K.A. An internal ribosome entry site in the 5′ untranslated region of epidermal growth factor receptor allows hypoxic expression. Oncogenesis 2015, 4, 134. [Google Scholar] [CrossRef]
- Komar, A.A.; Hatzoglou, M. Exploring Internal Ribosome Entry Sites as Therapeutic Targets. Front. Oncol. 2015. [Google Scholar] [CrossRef]
- SUZUKI, Y.; SHIRAI, K.; OKA, K.; MOBARAKI, A.; YOSHIDA, Y.; NODA, S.; OKAMOTO, M.; SUZUKI, Y.; ITOH, J.; ITOH, H.; et al. Higher pAkt Expression Predicts a Significant Worse Prognosis in Glioblastomas. J. Radiat. Res. 2010, 51, 343–348. [Google Scholar] [CrossRef]
- Wurth, L.; Gebauer, F. RNA-binding proteins, multifaceted translational regulators in cancer. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 881–886. [Google Scholar] [CrossRef]
- Walters, B.; Thompson, S.R. Cap-Independent Translational Control of Carcinogenesis. Front. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Bressler, K.R.; Thakor, N. Eukaryotic Initiation Factor 5B (eIF5B) Cooperates with eIF1A and eIF5 to Facilitate uORF2-Mediated Repression of ATF4 Translation. Int. J. Mol. Sci. 2018, 19, 4032. [Google Scholar] [CrossRef] [PubMed]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H.; et al. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene 2018, 37, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Sims, J.S.; Hornstein, N.; Mela, A.; Garcia, F.; Lei, L.; Gass, D.A.; Amendolara, B.; Bruce, J.N.; Canoll, P.; et al. Ribosome profiling reveals a cell-type-specific translational landscape in brain tumors. J. Neurosci. 2014, 34, 10924–10936. [Google Scholar] [CrossRef]
- Helmy, K.; Halliday, J.; Fomchenko, E.; Setty, M.; Pitter, K.; Hafemeister, C.; Holland, E.C. Identification of Global Alteration of Translational Regulation in Glioma In Vivo. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Wahba, A.; Rath, B.H.; Bisht, K.; Camphausen, K.; Tofilon, P.J. Polysome profiling links translational control to the radioresponse of glioblastoma stem-like cells. Cancer Res. 2016, 76, 3078–3087. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| eIFs and Modulators | Gene | REMBRANDT | TCGA | |||||
|---|---|---|---|---|---|---|---|---|
| Oligodendroglioma | Astrocytoma | GBM | Classical | Mesenchymal | Neural | Proneural | ||
| eIF1 | EIF1 | ns | ns | ns | ns | ns | ns | ns |
| eIF1A | EIF1A | ns | ns | ns | ns | ns | ns | ns |
| eIF1AD | EIF1AD | + + | ns | + + + | ns | ns | ns | ns |
| eIF2 alpha | EIF2S1 | ns | ns | ns | ns | ns | ns | ns |
| eIF2 beta | EIF2S2 | + + | + | + + + | ns | ns | ns | ns |
| eIF2 gamma | EIF2S3 | + + + | + + | + + + | ns | + | ns | ns |
| HRI | EIF2AK1 | + + | + | + + | ns | ns | ns | ns |
| PKR | EIF2AK2 | + | + + | + | ns | ns | ns | ns |
| PERK | EIF2AK3 | ns | ns | + + + | ns | ns | ns | ns |
| GCN2 | EIF2AK4 | ns | ns | + + | ns | ns | ns | ns |
| eIF2B1 | EIF2B1 | + + + | + + + | + + + | ns | ns | ns | + + |
| eIF2B2 | EIF2B2 | + + + | + + + | + + + | ns | ns | ns | ns |
| eIF2B3 | EIF2B3 | − − − | − − − | ns | − | ns | ns | ns |
| eIF2B4 | EIF2B4 | + + + | + + + | + + + | ns | ns | + | ns |
| eIF2B5 | EIF2B5 | + + | + | + + | ns | ns | ns | ns |
| eIF3A | EIF3A | + + + | + + + | ns | ns | ns | ns | ns |
| eIF3B | EIF3B | + + + | + + + | + + + | + + + | + + + | ns | + + |
| eIF3D | EIF3D | + + + | + + + | + + + | + | + + | ns | + + |
| eIF3E | EIF3E | + + + | + + + | + + + | ns | ns | ns | ns |
| eIF3F | EIF3F | + + + | + + + | + + + | ns | ns | ns | ns |
| eIF3G | EIF3G | + + + | + + + | + + + | + + + | + + | + + | + + |
| eIF3H | EIF3H | + + + | + + + | + + + | ns | ns | ns | ns |
| eIF3I | EIF3I | ns | ns | + + + | + | + | + + + | ns |
| eIF3J | EIF3J | + | ns | ns | ns | ns | ns | ns |
| eIF3K | EIF3K | ns | ns | ns | ns | ns | + | ns |
| eIF3L | EIF3L | + + + | + + + | ns | ns | ns | ns | ns |
| eIF3M | EIF3M | + + + | + + | + + + | ns | ns | ns | ns |
| eIF4A1 | EIF4A1 | + + + | + + + | + + + | + + | + + + | + | + + + |
| eIF4A2 | EIF4A2 | − − − | − − − | − − − | − − − | − − − | ns | − − − |
| eIF4A3 | EIF4A3 | + + + | + + + | + + + | + | + + | + + + | + + |
| eIF4B | EIF4B | + + | ns | ns | ns | ns | ns | ns |
| eIF4E1 | EIF4E1 | − − − | − − − | − − − | − − − | − − − | ns | − − − |
| eIF4E2 | EIF4E2 | + + | + + | + + + | + + | + + + | + + + | + |
| eIF4E3 | EIF4E3 | − − − | − − − | − − − | − − − | − − − | ns | − − − |
| 4E-BP1 | EIF4EBP1 | + + + | + + + | + + + | + | + + + | + + | + + + |
| 4E-BP2 | EIF4EBP2 | + + + | + + + | ns | + | + | + | ns |
| 4E-BP3 | ANKHD1 | + + + | + + + | + + + | ns | ns | ns | ns |
| 4E-T | EIF4ENIF1 | ns | − | − − − | − − − | − − − | − − − | − − |
| eIF4G1 | EIF4G1 | + + + | + + + | + + + | ns | ns | ns | ns |
| eIF4G2 | EIF4G2 | + + + | ns | + + + | ns | ns | ns | ns |
| eIF4G3 | EIF4G3 | − − − | − − − | − − − | − − − | − − − | − − − | − − |
| eIF4H | EIF4H | ns | ns | ns | ns | ns | − | ns |
| PDCD4 | PDCD4 | ns | ns | ns | − − | ns | ns | ns |
| eIF5 | EIF5 | ns | − − | ns | ns | − | ns | − − |
| eIF5B | EIF5B | + + + | + + + | + + + | ns | ns | ns | ns |
| eIF6 | EIF6 | + + + | + + | + + + | + + | + + | + + | ns |
| ITAFs | Gene | REMBRANDT | TCGA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Oligodendroglioma | Astrocytoma | GBM | Classical | Mesenchymal | Neural | Proneural | Activity | ||
| Class I | |||||||||
| Annexin A2 | ANXA2 | ns | + + | + + + | + + + | + + + | + | ns | A |
| CUGBP1 | CUGBP1 | ns | ns | ns | ns | − − | − − − | ns | A/I |
| DAP5 # | EIF4G2 | + + + | ns | + + + | ns | ns | ns | ns | A |
| FBP3 | FUBP3 | + + + | + + + | + + + | + + + | + + + | + | + + + | A |
| FUS | FUS | + + + | + + + | + + + | + + | ns | ns | + + + | A |
| GRSF1 | GRSF1 | − − − | − − − | − − − | − − | − − − | ns | − | A |
| H-ferritin | FTH1 | ns | ns | ns | ns | ns | ns | ns | A |
| HDMX | MDM4 | + + + | + + + | + + + | ns | ns | ns | + + | A |
| hnRNPA1 | HNRNPA1 | + + + | + + | ns | + + | ns | ns | + + + | A/I |
| hnRNPC | HNRNPC | + + + | + + + | + + + | ns | ns | ns | ns | A |
| hnRNPD | HNRNPD | + + + | + + + | + | ns | ns | ns | ns | A |
| hnRNPE1 | PCBP1 | ns | ns | + + | ns | ns | ns | ns | A |
| hnRNPE2 | PCBP2 | + + + | + + + | + + + | + | ns | ns | + + + | A |
| hnRNPE3 | PCBP3 | ns | ns | − − − | − − − | − − − | − − | − − | A |
| hnRNPE4 | PCBP4 | + + | ns | ns | ns | ns | ns | + + + | A |
| hnRNPH2 | HNRNPH2 | ns | ns | − − − | − − − | − − | − − | − − | A |
| hnRNPK | HNRNPK | + + + | + + + | + + + | + + | ns | ns | + + + | A |
| hnRNPL | HNRNPL | ns | ns | ns | ns | ns | ns | + + + | A |
| hnRNPM | HNRNPM | + + + | + + + | + + + | + + | ns | ns | ns | A |
| hnRNPQ | SYNCRIP | + + + | + + + | + + + | + | ns | ns | + + | A |
| hnRNPR | HNRNPR | + + | + + + | + + + | ns | ns | ns | + | A |
| HuR | ELAV1 | + | + | + + + | − − | − − − | − − − | ns | A/I |
| La auto antigen | SSB | + | + | + + | ns | ns | ns | ns | A/I |
| Mdm2 | MDM2 | + + + | + + + | + + + | ns | + | + | ns | A |
| NF45 | ILF2 | + + + | + + + | + + + | + + | ns | + + + | + + + | A |
| nPTB | PTBP2 | − − − | − − | − − − | − − − | − − − | ns | ns | A |
| nucleolin | NCL | Not available | ns | ns | ns | ns | A/I | ||
| p54nrb | NONO | + + + | + + + | + + + | + + + | + + | ns | + + + | A |
| PDCD4 # | PDCD4 | ns | ns | ns | − − | ns | ns | ns | A/I |
| PSF | SFPQ | + + | ns | ns | ns | ns | ns | + | A/I |
| PTB | PTBP1 | + + + | + + + | + + + | + + + | + + + | ns | + + + | A/I |
| RHA | DHX9 | + + + | + + + | + + + | ns | ns | ns | + | A |
| SMAR 1 | BANP | + + + | + + + | + + + | + | + + | + + + | + + | A/I |
| YB1 | YBX1 | + + + | + + + | + + + | + + + | + + | + | + + + | A |
| Class II | |||||||||
| 4E-BP1 # | EIF4EBP1 | + + + | + + + | + + + | + | + + + | + + | + + + | A |
| APP (AICD) | APP | − − − | − − − | − − − | − − | − − | − − − | − − − | A |
| eeF1A2 | EEF1A2 | − − − | − − − | − − − | − − − | − − − | − | − | A |
| eIF3A # | EIF3A | + + + | + + + | ns | ns | ns | ns | ns | A |
| eIF3B # | EIF3B | + + + | + + + | + + + | + + + | + + + | ns | + + | A |
| eIF3D # | EIF3D | + + + | + + + | + + + | + | + + | ns | + + | A |
| eIF3E # | EIF3E | + + + | + + + | + + + | ns | ns | ns | ns | A |
| eIF3F # | EIF3F | + + + | + + + | + + + | ns | ns | ns | ns | A |
| eIF3G # | EIF3G | + + + | + + + | + + + | + + + | + + | + + | + + | A |
| eIF3H # | EIF3H | + + + | + + + | + + + | ns | ns | ns | ns | A |
| eIF3I # | EIF3I | ns | ns | + + + | + | + | + + + | ns | A |
| eIF3J # | EIF3J | + | ns | ns | ns | ns | ns | ns | A |
| eIF3K # | EIF3K | ns | ns | ns | ns | ns | + | ns | A |
| eIF3L # | EIF3L | + + + | + + + | ns | ns | ns | ns | ns | A |
| eIF3M # | EIF3M | + + + | + + | + + + | ns | ns | ns | ns | A |
| eIF4A1 # | EIF4A1 | + + + | + + + | + + + | + + | + + + | + | + + + | A |
| eIF4A2 # | EIF4A2 | − − − | − − − | − − − | − − − | − − − | ns | − − − | A |
| eIF4A3 # | EIF4A3 | + + + | + + + | + + + | + | + + | + + + | + + | A |
| eIF4G1 # | EIF4G1 | + + + | + + + | + + + | ns | ns | ns | ns | A |
| eIF5B # | EIF5B | + + + | + + + | + + + | ns | ns | ns | ns | A |
| eL38 | RPL38 | + + + | + + + | + + | − | ns | ns | ns | A |
| eS19 | RPS19 | + + + | + + + | + + + | + + + | + + + | + + + | + + | A |
| eS25 | RPS25 | + + + | + + + | + + + | ns | ns | + + | ns | A |
| Gemin5 | GEMIN5 | + + + | + + + | + + + | + | + | ns | + + | A/I |
| Hepsin | HPN | − − − | − − − | − | ns | ns | ns | ns | I |
| PINK1 | PINK1 | − − − | − − − | − − − | − − − | − − − | − − | − − − | A |
| Rack1 | GNB2L1 | + + + | + + + | + + + | + + + | + + + | + | + + + | A/I |
| TCP80 | ILF3 | + + + | + + + | + + + | + + + | ns | ns | + + + | A |
| uL1 | RPL10A | + + + | + + + | + + + | ns | ns | + + | ns | A |
| uL24 | RPL26 | Not available | ns | ns | ns | ns | A | ||
| uL5 | RPL11 | + + + | + + + | + + + | ns | + + + | + | + + + | A |
| VASH1 | VASH1 | + | + + | + | + + | ns | ns | + + | A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marina, D.; Arnaud, L.; Paul Noel, L.; Felix, S.; Bernard, R.; Natacha, C. Relevance of Translation Initiation in Diffuse Glioma Biology and its Therapeutic Potential. Cells 2019, 8, 1542. https://doi.org/10.3390/cells8121542
Marina D, Arnaud L, Paul Noel L, Felix S, Bernard R, Natacha C. Relevance of Translation Initiation in Diffuse Glioma Biology and its Therapeutic Potential. Cells. 2019; 8(12):1542. https://doi.org/10.3390/cells8121542
Chicago/Turabian StyleMarina, Digregorio, Lombard Arnaud, Lumapat Paul Noel, Scholtes Felix, Rogister Bernard, and Coppieters Natacha. 2019. "Relevance of Translation Initiation in Diffuse Glioma Biology and its Therapeutic Potential" Cells 8, no. 12: 1542. https://doi.org/10.3390/cells8121542
APA StyleMarina, D., Arnaud, L., Paul Noel, L., Felix, S., Bernard, R., & Natacha, C. (2019). Relevance of Translation Initiation in Diffuse Glioma Biology and its Therapeutic Potential. Cells, 8(12), 1542. https://doi.org/10.3390/cells8121542