Effect of Notch and PARP Pathways’ Inhibition in Leukemic Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Growth Curve and Treatment
2.3. RNA Isolation and Preparation of cDNA
2.4. Quantitative Real-Time PCR
2.5. Western Blot
2.6. Statistical Methods
3. Results
3.1. Effect of Notch and PARP inhibition on Jurkat, CLL and 697 Cells’ Proliferation
3.2. Effect of Notch and PARP Inhibition on Jurkat, CLL and 697 Cells’ mRNA Expression
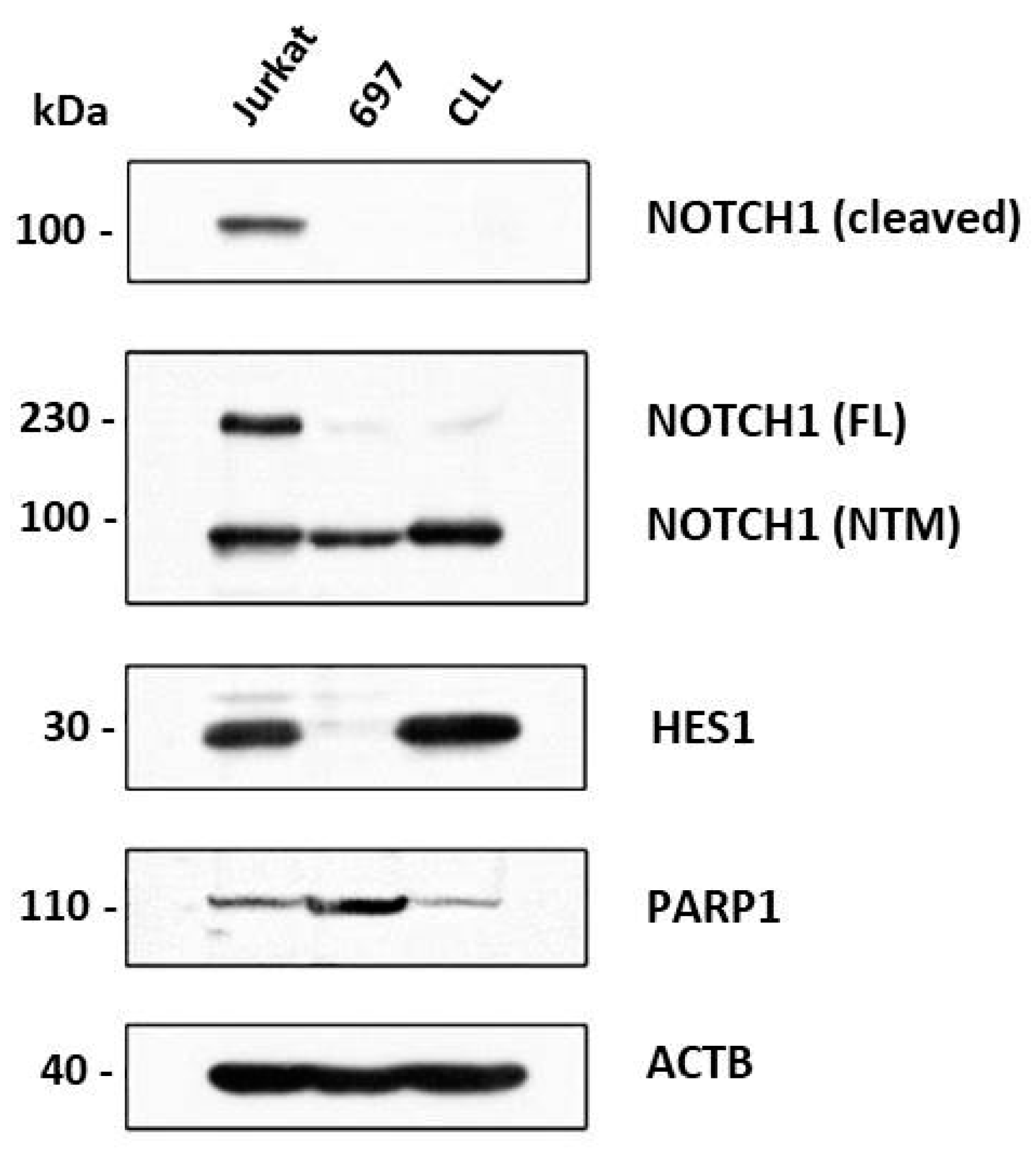
3.3. Effect of Notch and PARP Inhibition on Jurkat, CLL and 697 Cells’ Protein Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.S.; Radtke, F. Notch as a tumor suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, L.; Comi, P.; Cobos, E.; Martin Kast, W.; Chiriva-Internati, M.; Chiaramonte, R. Notch-ing from T-cell to B-cell lymphoid malignancies. Cancer Lett. 2011, 308, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Kraus, W.L. The PARP Side of the Nucleus: Molecular Actions, Physiological Outcomes, and Clinical Targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.G.; Solum, D.; Song, E.J.; Lee, K.-J.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. Activating the PARP1 Sensor Component of the Groucho/TLE1 Corepressor Complex Mediates a CaMKinaseIIδ-Dependent Neurogenic Gene Activation Pathway. Cell 2004, 119, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Fang, W.; Song, G.; Mullighan, C.G.; Hammitt, R.; McMurray, J.; Zweidler-McKay, P.A. Notch/HES1-mediated PARP1 activation: A cell type-specific mechanism for tumor suppression. Blood 2011, 117, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Primer3Plus. Available online: http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi (accessed on 15 May 2016).
- PrimerQuest Tool|IDT. Available online: https://eu.idtdna.com/Primerquest/Home/Index (accessed on 20 May 2016).
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.F.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell. Mol. Life Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef] [PubMed]
- Garcia Soriano, F.; Virag, L.; Jagtap, P.; Szabo, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly (ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Okuhashi, Y.; Nara, N.; Tohda, S. Effects of gamma-secretase inhibitors on the growth of leukemia cells. Anticancer Res. 2010, 30, 495–498. [Google Scholar] [PubMed]
- Dumortier, A.; Jeannet, R.; Kirstetter, P.; Kleinmann, E.; Sellars, M.; dos Santos, N.R.; Thibault, C.; Barths, J.; Ghysdael, J.; Punt, J.A.; et al. Notch activation is an early and critical event during T-Cell leukemogenesis in Ikaros-deficient mice. Mol. Cell. Biol. 2006, 26, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Henkel, T.; Ling, P.D.; Hayward, S.D.; Peterson, M.G. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 1994, 265, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Sulis, M.L.; Williams, O.; Palomero, T.; Tosello, V.; Pallikuppam, S.; Real, P.J.; Barnes, K.; Zuurbier, L.; Meijerink, J.P.; Ferrando, A.A. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood 2008, 112, 733–740. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, M.J.; Selkoe, D.J. The Notch Ligands, Jagged and Delta, Are Sequentially Processed by α-Secretase and Presenilin/γ-Secretase and Release Signaling Fragments. J. Biol. Chem. 2003, 278, 34427–34437. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Hales, E.C.; Orr, S.M.; Larson Gedman, A.; Taub, J.W.; Matherly, L.H. Notch1 receptor regulates AKT protein activation loop (Thr308) dephosphorylation through modulation of the PP2A phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell acute lymphoblastic leukemia cells. J. Biol. Chem. 2013, 288, 22836–22848. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.; Bhatnagar, H.; Lin, A.P.; Wang, L.; Aster, J.C.; Sill, H.; Aguiar, R.C. A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia 2014, 29, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Pathak, S.; Mandal, M.; Trinh, L.; Clark, M.R.; Lu, R. IKZF1 and IKZF3 Inhibit Pre-B-Cell Proliferation by Directly Suppressing c-MYC Expression. Mol. Cell. Biol. 2010, 30, 4149–4158. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine Factors Deployed by Tumor Vascular Niche Induce B Cell Lymphoma Invasiveness and Chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.C.; Klobucar, K.; Pierre, C.C.; Ansari, A.; Zhenilo, S.; Prokhortchouk, E.; Daniel, J.M. Kaiso differentially regulates components of the Notch signaling pathway in intestinal cells. Cell Commun. Signal. 2017, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Crotty, M.L.; Sensel, M.; Sather, H.; Navara, C.; Nachman, J.; Steinherz, P.G.; Gaynon, P.S.; Seibel, N.; Mao, C.; et al. Expression of dominant-negative IKZF1 isoforms in T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 1999, 5, 2112–2120. [Google Scholar] [PubMed]
- Oestreich, K.J.; Weinmann, A.S. IKZF1 changes the face of NuRD remodeling. Nat. Immunol. 2011, 13, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Billot, K.; Soeur, J.; Chereau, F.; Arrouss, I.; Merle-Béral, H.; Huang, M.E.; Mazier, D.; Baud, V.; Rebollo, A. Deregulation of IKZF3 expression in chronic lymphocytic leukemia is associated with epigenetic modifications. Blood 2011, 117, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Jeannet, R.; Mastio, J.; Macias-Garcia, A.; Oravecz, A.; Ashworth, T.; Geimer Le Lay, A.S.; Jost, B.; Le Gras, S.; Ghysdael, J.; Gridley, T.; et al. Oncogenic activation of the Notch1 gene by deletion of its promoter in IKZF1-deficient T-ALL. Blood 2010, 116, 5443–5454. [Google Scholar] [CrossRef] [PubMed]
- Kathrein, K.L.; Chari, S.; Winandy, S. IKZF1 directly represses the notch target gene Hes1 in a leukemia T cell line: Implications for CD4 regulation. J. Biol. Chem. 2008, 283, 10476–10484. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.T.; Cimmino, L.; Hu, Y.; Trimarchi, T.; Tagoh, H.; McKenzie, M.D.; Best, S.A.; Tuohey, L.; Willson, T.A.; Nutt, S.L.; et al. Activated Notch Counteracts Ikaros Tumor Suppression in Mouse and Human T-Cell Acute Lymphoblastic Leukemia. Leukemia 2015, 29, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, K.; Kondo, H.; Okamura, Y.; Shima, H.; Kurokochi, Y.; Kimura, K.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yui, K.; et al. Zinc finger-IRF composite elements bound by Ikaros/IRF4 complexes function as gene repression in plasma cell. Blood Adv. 2018, 2, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Marke, R.; van Leeuwen, F.N.; Scheijen, B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 103, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Cappellini, A.; Oliveira, M.; Fragoso, R.; Barata, J.T.; Bertaina, A.; Locatelli, F.; Simioni, C.; Neri, L.M.; Chiarini, F.; et al. Phosphatidylinositol 3-kinase inhibition potentiates glucocorticoid response in B-cell acute lymphoblastic leukemia. J. Cell. Physiol. 2018, 233, 1796–1811. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Landhuis, E.; Dose, M.; Hazan, I.; Zhang, J.; Naito, T.; Jackson, A.F.; Wu, J.; Perotti, E.A.; Kaufmann, C.; et al. Transcriptional Regulation of the Ikzf1 Locus. Blood 2013, 122, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, A.; Duhamel, M.; Fleischer, A.; Reimann, A.; Dessauge, F.; Rebollo, A. Critical function of IKZF1 in controlling IKZF3 gene expression. FEBS Lett. 2007, 581, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Rosati, E.; Sabatini, R.; Rampino, G.; Tabilio, A.; Di Ianni, M.; Fettucciari, K.; Bartoli, A.; Coaccioli, S.; Screpanti, I.; Marconi, P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood 2009, 113, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Schreiber, V.; Saenz, L.; Martínez, T.; Muñoz-Suano, A.; Dominguez-Villar, M.; Ramírez, P.; Parrilla, P.; Ag uado, E.; García-Cózar, F.; et al. Regulation of NFAT by poly(ADP-Ribose) Polymerase Activity in T Cells. Mol. Immunol. 2008, 45, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. A Role of Poly (ADP-Ribose) Polymerase in NF-κB Transcriptional Activation. Biol. Chem. 1999, 380, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Kang, Y.; Massagué, J. Defective repression of c-MYC in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proc. Natl. Acad. Sci. USA 2001, 98, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Lönn, P.; van der Heide, L.P.; Dahl, M.; Hellman, U.; Heldin, C.H.; Moustakas, A. PARP1 attenuates Smad-mediated transcription. Mol. Cell 2010, 40, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Beck, S.; Pollard, S.M. Differentiation therapy for glioblastoma—Too many obstacles? Mol. Cell. Oncol. 2016, 3, e1124174. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Bhat, S.A.; Gogoi, D.; Gokhale, A.; Chiplunkar, S.V. Inhibition of Notch signalling has ability to alter the proximal and distal TCR signalling events in human CD3 + αβ T-cells. Mol. Immunol. 2017, 92, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Matlawska-Wasowska, K.; Girodon, F.; Mazel, T.; Willman, C.L.; Atlas, S.; Chen, I.M.; Harvey, R.C.; Hunger, S.P.; Ness, S.A.; et al. GSI-I (Z-LLNle-CHO) inhibits γ-secretase and the proteosome to trigger cell death in precursor-B acute lymphoblastic leukemia. Leukemia. 2011, 25, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Kogoshi, H.; Sato, T.; Koyama, T.; Nara, N.; Tohda, S. Gamma-secretase inhibitors suppress the growth of leukemia and lymphoma cells. Oncol. Rep. 2007, 18, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.A.; Cesaroni, M.; Denny, M.F.; Lupey, L.N.; Tempera, I. Global Transcriptome Analysis Reveals That Poly(ADP-Ribose) Polymerase 1 Regulates Gene Expression through EZH2. Mol. Cell. Biol. 2015, 35, 3934–3944. [Google Scholar] [CrossRef] [PubMed]
- Weston, V.J.; Oldreive, C.E.; Skowronska, A.; Oscier, D.G.; Pratt, G.; Dyer, M.J.S.; Smith, G.; Powell, J.E.; Rudzki, Z.; Kearns, P.; et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horvat, L.; Antica, M.; Matulić, M. Effect of Notch and PARP Pathways’ Inhibition in Leukemic Cells. Cells 2018, 7, 58. https://doi.org/10.3390/cells7060058
Horvat L, Antica M, Matulić M. Effect of Notch and PARP Pathways’ Inhibition in Leukemic Cells. Cells. 2018; 7(6):58. https://doi.org/10.3390/cells7060058
Chicago/Turabian StyleHorvat, Luka, Mariastefania Antica, and Maja Matulić. 2018. "Effect of Notch and PARP Pathways’ Inhibition in Leukemic Cells" Cells 7, no. 6: 58. https://doi.org/10.3390/cells7060058
APA StyleHorvat, L., Antica, M., & Matulić, M. (2018). Effect of Notch and PARP Pathways’ Inhibition in Leukemic Cells. Cells, 7(6), 58. https://doi.org/10.3390/cells7060058